Onkologia
Nowotwory spowodowane są przez uszkodzenia komórek w organizmie i stanowią ważny problem zdrowotny.
Onkologia
Dowiedz się więcej na temat raka prostaty, raka pęcherza moczowego, raka nerki i nowotworów neuroendokrynnych.
Nowotwory Neuroendokrynne
Nowotwory neuroendokrynne (NET) występują najczęściej w przewodzie pokarmowym. Czasami nie wykazują żadnych objawów, mogą również produkować nieprawidłowo duże ilości hormonów, które wpływają na funkcjonowanie organizmu, a także powodować problemy trawienne, utratę masy ciała oraz zaczerwienienie skóry twarzy i napady gorąca. Dowiedz się więcej na temat tego rzadkiego zaburzenia endokrynologicznego, jego diagnozowania i różnych metod leczenia.
Definicja
Komórki endokrynne są obecne w całym organizmie i w związku z tym, nowotwory neuroendokrynne (NET) mogą powstawać w wielu narządach. W około 60% przypadków NET pojawiają się w układzie pokarmowym (GEP NET). Mogą występować w przewodzie pokarmowym od przełyku do odbytnicy poprzez okrężnicę i trzustkę. Pierwotne nowotwory mogą rozprzestrzeniać się do innych narządów, zwłaszcza do wątroby.
Komórki nowotworowe NET wykazują cechy wspólne dla komórek nerwowych i wewnątrzwydzielniczych (takie jak zdolność do wydzielania hormonów), stąd nazwa „nowotwory neuroendokrynne”.
Objawy i skutki zdrowotne
Wiele nowotworów neuroendokrynnych nie wykazuje żadnych objawów klinicznych ze względu na swoje niewielkie rozmiary i brak wydzielania hormonów. Jeśli powodują objawy, są one różnorodne w zależności od narządu w jakim się rozwijają i manifestują się powolnie w ciągu kilku lat.
Objawy powodowane przez wzrost guza
Mimo, że nowotwory neuroendokrynne przewodu pokarmowego składają się z komórek pochodzenia hormonalnego, większość z nich nie powoduje nadmiernego wydzielania hormonów, a zatem nie wykazuje objawów klinicznych. Guzy te, zwane „nieczynnymi”, zostają wykryte dopiero wtedy, gdy osiągają znaczne rozmiary i powodują różne objawy. Są to głównie:
- bóle lub dyskomfort brzucha,
- wzdęcia,
- zaparcia,
- niedrożność jelit,
- krew w stolcu,
- nudności lub wymioty,
- utrata masy ciała,
- żółtaczka.
Objawy powodowane przez guza wytwarzającego hormony
Niektóre GEP NET wydzielają nadmiar hormonów odpowiedzialnych za różne zaburzenia:
- biegunkę,
- utratę masy ciała,
- odwodnienie,
- zaczerwienienie skóry i uderzenia gorąca,
- świszczący oddech,
- palpitacje serca.
Czynniki ryzyka
Większość chorych z nowotworami neuroendokrynnymi nie ma żadnych zidentyfikowanych czynników ryzyka. W rzadkich przypadkach NET przewodu pokarmowego są częścią choroby dziedzicznej i mamy wówczas do czynienia z zespołem predyspozycji genetycznej, przy możliwym współwystępowaniu innych nowotworów. Najczęstszym z tych zespołów jest zespół mnogiej gruczolakowatości wewnątrzwydzielniczej typu 1 (MEN 1), który obejmuje głównie guzy trzustki, przysadki, przytarczyc i nadnerczy. Nie ma metod zapobiegających powstawaniu NET.
Częstość występowania
Nowotwory neuroendokrynne (NET) są rzadkie, odnotowuje się od 2 do 5 nowych przypadków rocznie na 100 000 osób. Są one częstsze u osób w starszym wieku niż u osób młodych, ze średnią wieku w momencie rozpoznania około 65 lat, i występują zarówno u mężczyzn, jak i kobiet.
Ze względu na brak objawów lub obecność niespecyficznych objawów choroby, GEP NET mogą pozostawać przez wiele lat niezauważone. Rozpoznanie następuje często w późnym stadium choroby, z opóźnieniem średnio od 5 do 7 lat. Dlatego też w 20 do 50% przypadków w momencie wykrycia, choroba jest już rozprzestrzeniona.
Dostęnych jest kilka badań umożliwiających rozpoznanie GEP NET:
- Ultrasonografia endoskopowa (endosonografia-badanie obrazowe za pomocą ultradźwięków) pozwala zobrazować guzy znajdujące się wewnątrz przewodu pokarmowego oraz wykonać biopsję.
- Tomografia komputerowa (CT) i rezonans magnetyczny (MRI) umożliwiają uwidocznienie guzów, ale nie mogą potwierdzić ich endokrynnego charakteru.
- Badania medycyny nuklearnej polegające na iniekcji markerów, które przyłączają się do receptorów NET (scyntygrafia, pozytonowa tomografia emisyjna połączona z tomografią komputerową) potwierdzają charakter endokrynny guza.
- Badania krwi pozwalają wykryć czynne guzy NET z powodu nieprawidłowego wzrostu poziomu wydzielanych przez nie hormonów.
- Badanie mikroskopowe guza lub pobranej próbki jest jedynym badaniem umożliwiającym potwierdzenie rozpoznania. W tym celu wykonuje się często biopsję guza, czyli pobiera się niewielki fragment do badania mikroskopowego (histopatologicznego).
Ze względu na różnorodność oraz powolną ewolucję, okresy remisji i nawrotów przez bardzo długi czas (kilka lat lub nawet dekad) w przypadku NET stosuje się różne metody leczenia. Pacjent z guzem neuroendokrynnym może być poddany różnym rodzajom leczenia.
Główne opcje leczenia:
- Interwencja chirurgiczna, która pozwala usunąć guz lub zmniejszyć objętość guzów, jeśli występują w postaci rozsianej. Opcja chirurgiczna powinna być rozpatrywana przez interdyscyplinarne konsylium.
- Chemioterapia, podawana w sposób przerywany, w cyklach, w celu zniszczenia komórek nowotworowych. Stosowana jest indywidualne lub w połączeniu z zabiegiem chirurgicznym.
- Embolizacja (zamknięcie tętnicy w celu zatrzymania dopływu krwi do guza, aby spowodować jego martwicę) oraz fale o częstotliwości radiowej są stosowane bardzo rzadko, głównie podczas miejscowych zabiegów chirurgicznych wątroby.
- Analogi somatostatyny (syntetyczne odpowiedniki hormonu somatostatyny) stosowane często w sposób ciągły.
- Terapie celowane.
- Radioterapia wewnętrzna, która wiąże się z receptorami somatostatyny.
W niektórych przypadkach, gdy nie ma objawów, a guz nie rośnie (lub rośnie bardzo powoli) i nie może być całkowicie usunięty, pacjenci nie są poddawani żadnym zabiegom medycznym ani chirurgicznym i są obserwowani przy pomocy badań biologicznych, radiologicznych i klinicznych. Bez względu na to, czy guz jest leczony czy nie, pacjenci wymagają regularnej kontroli różnych specjalistów w specjalistycznych ośrodkach opieki zdrowotnej zajmujących się leczeniem NET.
5,25 / 100 000 / rok
Około 60% nowotworów neuroendokrynnych to nowotwory żołądkowo-jelitowo-trzustkowe2
Około 65 lat
Średni wiek w chwili rozpoznania 3
Źródła
[1]Kos-Kudła et al. Zalecenia ogólne dotyczące postępowania diagnostyczno-terapeutycznego w nowotworach neuroendokrynnych układu pokarmowego (rekomendowane przez Polską Sieć Guzów Neuroendokrynnych) Endokrynol Pol;2017; 68(2);79-110
[2]Frilling A et al. Neuroendocrine tumor disease: an evolving landscape; Endocr Relat Cancer 2012;19:R163-R185
[3] Lepage C, Bouvier AM, Phelip JM, Hatem C, Vernet C, Faivre J. Incidence and management of malignant digestive endocrine tumours in a well defined French population. Gut 2004; 53(4):549-53
Rak nerki
Rak nerki (ang. renal cell carcinoma, RCC) stanowi 2–3% wszystkich nowotworów złośliwych. Poniżej znajdują się informacje na temat tej choroby, jej rozpoznania i metod leczenia.
Objawy
Do przedmiotowych i podmiotowych objawów raka nerki należą:
- obecność krwi w moczu;
- stale utrzymujący się ból w boku, tuż poniżej linii żeber;
- obecność wyczuwalnego guza w okolicy nerki (po którejkolwiek stronie ciała).
W przypadku wystąpienia któregokolwiek z tych objawów należy możliwie najszybciej zgłosić się do lekarza podstawowej opieki zdrowotnej.
W około połowie przypadków rak nerki nie daje żadnych objawów, a chorobę wykrywa się podczas badań wykonywanych w związku z innymi dolegliwościami.
Czynniki Ryzyka
Palenie papierosów, otyłość i nadciśnienie tętnicze są dobrze znanymi czynnikami ryzyka. Wydaje się także, że rak nerki częściej występuje u pacjentów dializowanych.
W około 2–3% przypadków rak nerki jest uwarunkowany genetycznie, przy czym opisano kilka zespołów dziedziczonych w sposób autosomalny dominujący, odrębnych genotypowo i fenotypowo1.
Częstosść występowania
Rak nerki stanowi 2–3% wszystkich nowotworów złośliwych2, przy czym częstość występowania tego nowotworu jest największa w krajach Europy zachodniej.
W czasie ostatnich dwudziestu lat notowano wzrost częstości występowania raka nerki o około 2% rocznie, zarówno na całym świecie, jak i w Europie, jedynie w Danii i Szwecji obserwowano stały spadek zachorowań3.
W 2012 roku stwierdzono około 84 400 nowych przypadków raka nerki i 34 700 zgonów z powodu raka nerki w krajach Unii Europejskiej4.
W Europie współczynnik śmiertelności całkowitej z powodu raka nerki wzrastał do wczesnych lat 90-tych XX wieku, a następnie ustabilizował się lub zaczął się obniżać. Spadek śmiertelności notuje się od lat 80-tych XX wieku w krajach skandynawskich i od początku lat 90-tych XX wieku we Francji, Niemczech, Austrii, Holandii oraz we Włoszech. Jednak w niektórych krajach Europy (Chorwacji, Estonii, Grecji, Irlandii, Słowacji) współczynnik śmiertelności wciąż wykazuje tendencję wzrostową5.
W wielu przypadkach guz w nerce pozostaje bezobjawowy aż do późnych stadiów zaawansowania choroby.
Obecnie w ponad 50% przypadków rak nerki jest wykrywany przy okazji nieinwazyjnych badań obrazowych wykonywanych z powodu różnych nieswoistych objawów i innych chorób jamy brzusznej6. Klasyczna triada objawów w postaci bólu w okolicy lędźwiowej, intensywnego krwiomoczu i wyczuwalnego w badaniu palpacyjnym guza w jamie brzusznej, jest obecnie rzadkim zjawiskiem (6–10%) i koreluje z zaawansowanym stadium choroby.
Inne objawy mogące towarzyszyć rakowi nerki stwierdza się u około 30% pacjentów. Do najczęściej występujących zalicza się: nadciśnienie tętnicze, wyniszczenie, utratę masy ciała, gorączkę, neuromiopatię, amyloidozę, wzrost wartości opadu krwinek czerwonych (OB), niedokrwistość, zaburzenia czynności wątroby, hiperkalcemię i policytemię.
Badanie fizykalne ma ograniczone znaczenie w rozpoznawaniu raka nerki. Jednak następujące objawy powinny stanowić wskazanie do wykonania badań radiologicznych: wyczuwalny palpacyjnie guz w jamie brzusznej; wyczuwalne palpacyjnie powiększenie szyjnych węzłów chłonnych; niezmniejszające się żylaki powrózka nasiennego oraz obustronny obrzęk kończyn dolnych, wskazujący na zajęcie naczyń żylnych7.
Jeśli objawy lub wyniki badania przedmiotowego wskazują na raka nerki, prawdopodobnie wykonane zostaną dodatkowe badania, w tym badania laboratoryjne i badania obrazowe.
- Badania laboratoryjne
- Badanie moczu
- Pełna morfologia krwi
- Badania biochemiczne krwi
- Badania obrazowe
- Tomografia komputerowa (TK), obrazowanie metodą rezonansu magnetycznego (ang. magnetic resonance imaging, MRI) i badania ultrasonograficzne mogą być bardzo pomocne w rozpoznaniu większości guzów nerki, chociaż rzadko u pacjentów konieczne jest wykonanie wszystkich tych badań.
- Inne badania, takie jak prześwietlenie (RTG) klatki piersiowej i scyntygrafia kości, wykonuje się częściej w celu ustalenia, czy nastąpił rozsiew nowotworu (przerzuty) do innych części ciała.
Dostępne metody leczenia :
- Leczenie chirurgiczne: w większości nowotworów nerki zabieg operacyjny jest podstawową metodą leczenia. Nawet u pacjentów, u których doszło do rozsiewu nowotworu do innych narządów, operacyjne usunięcie guza nerki może przynieść korzyść.
- Ablacja i inne metody leczenia miejscowego: niekiedy w celu zniszczenia guza nerki można zastosować następujące metody, chociaż nie zostały one uznane za leczenie standardowe: krioterapia (krioablacja), ablacja prądem o częstotliwości radiowej (ang. radiofrequency ablation, RFA), embolizacja naczyń tętniczych.
- Aktywny nadzór (obserwacja): pacjenci w podeszłym wieku lub o słabym stanie zdrowia, u których guz nerki ma niewielkie rozmiary (mniejszy niż 4 cm) mogą początkowo nie otrzymywać leczenia. Guzy u nich występujące, należy uważnie obserwować w celu kontroli, czy szybko się rozrastają lub powiększają do rozmiarów przekraczających 4 cm. Wówczas można je usunąć chirurgicznie lub stosować inne metody lecznicze.
- Radioterapia: radioterapię częściej stosuje się w celu złagodzenia lub zmniejszenia nasilenia objawów raka nerki.
- Terapia celowana: leki stosuje się w terapii pierwszego lub drugiego rzutu, w przypadku raka nerki w stadium zaawansowanym lub w stadium przerzutów. Mogą one często spowodować zmniejszenie masy guza lub zahamowanie wzrostu nowotworu na pewien czas, ale nie powodują wyleczenia raka nerki.
- Immunoterapia (terapia biologiczna): terapia biologiczna ma na celu wzmocnienie układu odpornościowego organizmu w celu ułatwienia zwalczenia lub zniszczenia komórek nowotworowych. W leczeniu można stosować cytokiny (interleukinę 2 [IL-2], interferon alfa) lub inhibitory immunologicznych punktów kontrolnych.
- Chemioterapia: w ramach chemioterapii stosuje się leki przeciwnowotworowe podawane dożylnie lub doustnie (w postaci tabletek). Leki te przenikają do krwi i docierają do wszystkich części ciała. Z tego względu chemioterapia może być użyteczna w leczeniu rozsianych nowotworów, tj. z przerzutami do innych narządów poza nerką.
2–3%
wszystkich nowotworów złośliwych
W wielu przypadkach guz w nerce pozostaje bezobjawowy
Leczeniem pierwszego wyboru jest leczenie operacyjne
Źródła :
1Chow WH, Dong LM, Devesa SS. . Epidemiology and risk factors for kidney cancer. Nat Rev Urol 2010; 7: 245–257
2European Network of Cancer Registries. Eurocim version 4.0. European incidence database V2.3, 730 entity dictionary (2001), Lyon, 2001.
3Lindblad P. Epidemiology of renal cell carcinoma. Scand J Surg 2004;93(2):88-96 http://www.ncbi.nlm.nih.gov/pubmed/15285559
4Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer 2013 Apr;49(6):1374-403. http://www.ncbi.nlm.nih.gov/pubmed/23485231
5Levi F, Ferlay J, Galeone C, et al. The changing pattern of kidney cancer incidence and mortality in Europe. BJU Int 2008 Apr;101(8):949-58 http://www.ncbi.nlm.nih.gov/pubmed/18241251
6Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up
7Guidelines on Renal Cell Carcinoma, European Association of Urology, 2014– Uroweb.org
8American Cancer Society. https://www.cancer.org/cancer/kidney-cancer
9European Association of Urology. Guidelines for Clear Cell Renal Cancers That Are Resistant to Vascular Endothelial Growth Factor Receptor–Targeted Therapy
Rak Pęcherza Moczowego
Rak pęcherza moczowego, szósty najczęstszy nowotwór u mężczyzn oraz dwunasty u kobiet na świecie, w roku 2012 był przyczyną 165 100 zgonów. Dowiedz się więcej na temat tej choroby, jej diagnozowania i różnych metod leczenia.
Definicja
Rak pęcherza moczowego obejmuje różne nowotwory powstające na wewnętrznej ścianie pęcherza (nabłonek dróg moczowych). Najczęstszym nowotworem pęcherza, odpowiedzialnym za 90–95% przypadków choroby, jest rak przejściowo- i płaskonabłonkowy.
Nowotwory pęcherza moczowego można podzielić na dwie kategorie:
- Powierzchowne nowotwory pęcherza (NMIBC, Non-Muscle Invasive Bladder Cancer): guz jest ograniczony do błony śluzowej pęcherza (urothelium) i nie nacieka mięśniówki ściany pęcherza moczowego. 70% przypadków raka pęcherza moczowego jest w chwili rozpoznania powierzchownym rakiem pęcherza moczowego. Powierzchowne nowotwory pęcherza moczowego dzielą się na trzy podtypy: Ta (70%), T1 (20%) oraz carcinoma in situ (Tis) (10%).
- Nowotwory naciekające mięśniówkę ściany pęcherza moczowego (MIBC, Muscle Invasive Bladder Cancer, postać naciekająca): nowotwór nacieka: ścianę pęcherza moczowego i może rozprzestrzeniać się na pobliskie narządy i/lub węzły chłonne. 30% przypadków raka pęcherza moczowego jest w chwili rozpoznania nowotworem naciekającym (T2, T4).
Objawy i skutki zdrowotne
Najczęstsze objawy raka pęcherza moczowego:
- obecność krwi w moczu (krwiomocz), najbardziej widoczny przejaw choroby, który powinien stać się podstawą do konsultacji z lekarzem;
- pieczenie podczas oddawania moczu;
- potrzeba bardzo częstego oddawania moczu (częstomocz) lub nieopanowana potrzeba oddawania moczu (nietrzymanie moczu);
- wrażenie niepełnego opróżnienia pęcherza po oddaniu moczu;
- ból brzucha lub kości;
- utrata masy ciała.
Czynniki ryzyka
Istnieje wiele czynników ryzyka wystąpienia raka pęcherza moczowego:
- palenie tytoniu: główny czynnik ryzyka — ryzyko wystąpienia raka pęcherza moczowego u palaczy jest od dwóch do sześciu razy wyższe w porównaniu z osobami niepalącymi;
- narażenie zawodowe na czynniki rakotwórcze, takie jak aminy aromatyczne i wielopierścieniowe węglowodory aromatyczne (znajdują się na przykład w barwnikach, rozpuszczalnikach, farbach, produktach spalania, gumie i tekstyliach);
- płeć: mężczyźni są zagrożeni 2–3 razy bardziej niż kobiety;
- wiek: ryzyko zachorowania na raka pęcherza rośnie wraz z wiekiem, średni wiek w chwili rozpoznania wynosi około 70. lat;
- przewlekłe zakażenie dróg moczowych;
- schistosomatoza, znana również jako bilharcjoza, przewlekła choroba tropikalna spowodowana przez pasożyty;
- niektóre leki, takie jak te wykorzystywane przy chemioterapii (np. cyklofosfamid), które mogą znacząco zwiększyć ryzyko późniejszego wystąpienia raka pęcherza;
- wcześniejsza radioterapia w okolicy miednicy;
- wcześniejsze występowanie raka pęcherza moczowego lub występowanie tego schorzenia w rodzinie.
Częstość występowania
6. najczęstszy nowotwór u mężczyzn oraz 12. u kobiet. W 2015 roku roczną częstość występowania szacowano się na 10,1 przypadków na 100 000 mężczyzn oraz 2,5 przypadków na 100 000 kobiet.
Jest drugim najczęstszym rakiem układu moczowo-płciowego, po raku prostaty. Występuje dwa – trzy razy częściej u mężczyzn niż u kobiet, a częstość jego występowania zwiększa się wraz z wiekiem, szczególnie po 60. roku życia.
Aby zwiększyć powodzenie leczenia, ważne jest rozpoznanie raka pęcherza moczowego we wczesnym jego stadium. Diagnostyka raka pęcherza obejmuje następujące badania:
- Badanie bakteriologiczne moczu (posiew moczu): to podstawowe badanie wykonywane w celu potwierdzenia obecności zakażenia i krwi w moczu oraz zbadania ich przyczyny.
- Cytologia moczu: materiał do cytologii jest pobierany z moczu lub podczas płukania pęcherza; cytologia polega na badaniu komórek, które są naturalnie wydalane z moczem pacjenta, w celu wykrycia wszelkich nieprawidłowości.
- USG pęcherza moczowego: badanie pęcherza za pomocą ultradźwięków umożliwia wykrycie guza.
- Urografia dożylna (IVU): badanie rentgenowskie dróg moczowych wykonywane po dożylnym podaniu kontrastu jodowego. Badanie pozwala obejrzeć nerki i moczowody w celu wykrycia nowotworu w tych narządach.
Fiberoskopia pęcherza (cystoskopia): badanie wnętrza pęcherza za pomocą układu optycznego pozwala stwierdzić obecność polipa pęcherza oraz określić jego wygląd i lokalizację. Badanie może obejmować biopsję (pobranie małej części narządu lub tkanki do badania).
Dobór leczenia zależy od stadium choroby oraz ogólnego stanu zdrowia pacjenta:
- Przezcewkowa resekcja guza pęcherza moczowego (TURBT, transurethral resection of the bladder tumour): w przypadku nowotworów nienaciekających mięśniówki ściany pęcherza moczowego najpierw ogląda się i wycina wszystkie widoczne guzy przezcewkowo za pomocą cystoskopu (wziernika). Fluorescencja osiągana dzięki zastosowaniu światła niebieskiego, w połączeniu z cystoskopią z użyciem światła białego oraz TURT, umożliwia prawidłowe wykrywanie nowotworów złośliwych, a zwłaszcza carcinoma in situ (CIS). Tkanki pobrane podczas biopsji są następnie badane pod mikroskopem, co pozwala na określenie ryzyka nawrotu lub rozwoju choroby. Uzupełniające leczenie pooperacyjne obejmuje chemioterapię lub immunoterapię (BCG, Bacillus Calmette-Guerin) w postaci wlewek dopęcherzowych, jak również ścisłą kontrolę lekarską.
- Cystektomia (usunięcie pęcherza): u pacjentów z guzem naciekającym wykonuje się usunięcie węzłów chłonnych znajdujących się po obu stronach pęcherza wraz z częściowym lub całkowitym usunięciem pęcherza.
- Radioterapia: stosowana indywidualnie ma zazwyczaj cel paliatywny, jeśli nie można wykonać zabiegu chirurgicznego (w przypadku pacjentów osłabionych lub w podeszłym wieku).
- Chemioterapia: stosowana w przypadku, gdy guz przekracza granice pęcherza.
- Chemioradioterapia: technika łącząca umiarkowane dawki radioterapii oraz „lekką” chemioterapię, która pomaga uniknąć usunięcia pęcherza w niektórych przypadkach niewielkich guzów inwazyjnych.
Wykrywanie CIS za pomocą cystoskopii z użyciem światła białego (z lewej) oraz światła niebieskiego (po prawej)
Źródło: dr Dirk Zaak, Monachium, Niemcy
6. najczęstszy
nowotwór u mężczyzn
12. najczęstszy
nowotwór u kobiet na całym świecie
165 100
Zgonów na świecie w 2012 roku
Źródło: dr Dirk Zaak, Monachium, Niemcy
Urologia. Ilustrowany podręcznik dla studentów i stażystów. Redakcja naukowa: Tomasz Drewa, Kajetan Juszczak.PZWL Wydawnictwo Lekarskie, Warszawa 2018
Rak prostaty
Rak prostaty jest najczęstszą postacią raka u mężczyzn w wieku powyżej 50. lat. Po nowotworach płuc stanowi drugą przyczynę zgonów z powodu raka. Dowiedz się więcej na temat tej choroby, jej diagnozowania i różnych metod leczenia.
Definicja
Rak prostaty jest wynikiem patologicznego rozrostu komórek gruczołu krokowego, męskiego narządu płciowego wytwarzającego płyn nasienny, który wraz z plemnikami tworzy spermę. Męskie hormony płciowe, szczególnie testosteron, wpływają na rozwój tego nowotworu i namnażanie się komórek nowotworowych.
Objawy i skutki zdrowotne
Rozwój raka prostaty jest stosunkowo powolny. Wczesne etapy są zwykle bezobjawowe poza problemami pojawiającymi się podczas oddawania moczu, które przypisuje się powiększeniu prostaty ze względu na wiek. Może to być ignorowane przez wiele lat.
Istnieje jednakże kilka objawów urologicznych, sugerujących obecność raka gruczołu krokowego:
- częste oddawanie moczu,
- nietrzymanie moczu,
- zmniejszenie siły strumienia moczu,
- niemożność oddania moczu.
W późniejszym etapie choroby mogą pojawić się inne objawy, takie jak ból kości.
Czynniki ryzyka
Mimo, że przyczyny raka prostaty nie są znane, istnieje wiele czynników ryzyka, które wydają się mu sprzyjać:
- Wiek: rak prostaty jest niezwykle rzadki u mężczyzn przed 40 rokiem życia, najczęściej jest diagnozowany po ukończeniu 70 lat.
- Wywiad rodzinny: postać rodzinna, to znaczy, gdy istnieją co najmniej 2 przypadki raka prostaty u krewnych pierwszego stopnia (ojciec, brat) lub drugiego stopnia (dziadek, wujek), stanowi 20% przypadków, natomiast postać dziedziczna (co najmniej 3 przypadki raka prostaty u krewnych pierwszego lub drugiego stopnia) stanowi 5% przypadków raka prostaty.
- Pochodzenie etniczne i geograficzne: liczba przypadków raka prostaty jest znacznie wyższa w Afryce, Europie Północnej oraz Ameryce Północnej, a w krajach Azji Południowo-Wschodniej częstość jego występowania jest bardzo niska.
Częstość występowania
Szacuje się, że w Europie i Ameryce Północnej u jednego na sześciu mężczyzn w ciągu ich życia zostanie rozpoznany rak prostaty. Nowotwór ten jest przyczyną ponad 80 000 zgonów rocznie w Europie. Chociaż śmiertelność choroby ma tendencję spadkową (wskaźnik przeżycia 5 lat wynosi około 83,4%), częstość jej wystąpienia znacznie wzrasta, szczególnie ze względu na starzenie się społeczeństwa.
Wczesna diagnoza raka prostaty daje największe szanse na przeżycie pacjenta. Raka prostaty można wykryć, wykonując następujące badania:
- Badanie gruczołu krokowego per rectum (DRE, Digital Rectal Examination): lekarz wkłada palec w rękawiczce do odbytu, aby zbadać palpacyjnie prostatę, wyczuć guza lub zmiany o rozmiarze, kształcie lub konsystencji, które mogą wskazywać na raka w strefie obwodowej prostaty.
- Pomiar stężenia specyficznego antygenu prostaty (PSA) w próbce krwi: metoda pomaga wykryć nowotwór we wczesnych stadiach choroby, co pozwala na szybkie objęcie pacjenta leczeniem, niemniej jest obarczona ryzykiem fałszywie pozytywnego wyniku, co należy mieć na uwadze diagnozując pacjenta.
- Ultrasonografia transrektalna prostaty (TRUS, Transrectal Ultrasonography): w tym badaniu do odbytnicy wkłada się sondę ultradźwiękową w celu zobrazowania gruczołu krokowego.
- Biopsja (pobranie niewielkiej części narządu lub tkanki do badania) jest wykonywana w momencie podejrzenia raka, aby postawić rozpoznanie oraz ocenić agresywność nowotworu.
W rozpoznaniu należy uwzględniać różne etapy rozwoju raka prostaty. Wielkość guza, zajęcie lub nie węzłów chłonnych oraz obecność lub brak przerzutów do innych części ciała pozwalają określić stopień zaawansowania choroby, a tym samym — odpowiedni dobór leczenia.
Skuteczność leczenia raka prostaty w ostatnich latach uległa znacznej poprawie przy jednoczesnej, coraz większej indywidualizacji sposobu podejścia do pacjenta i jego leczenia. Bardziej indywidualne, a przy tym multidyscyplinarne podejście do terapii pozwala na lepsze
prowadzenie chorego na wszystkich etapach choroby.
W leczeniu raka gruczołu krokowego mogą być rozważane różne opcje terapeutyczne w zależności od stanu zdrowia pacjenta oraz stopnia zaawansowania choroby:
- Interwencja chirurgiczna (radykalna prostatektomia) to miejscowe leczenie raka, które polega na operacyjnym usunięciu całego gruczołu krokowego i pęcherzyków nasiennych. Często stosowana w niektórych zlokalizowanych nowotworach prostaty o podwyższonym ryzyku lub miejscowo zaawansowanych.
- Radioterapia zewnętrzna to miejscowe leczenie raka, które ma na celu zniszczenie komórek nowotworowych w obrębie gruczołu krokowego przy użyciu promieniowania. Radioterapię stosuje się jako samodzielne leczenie w przypadkach nowotworów zlokalizowanych niskiego i średniego ryzyka oraz w połączeniu z terapią hormonalną w przypadku raka prostaty wysokiego ryzyka i miejscowo zaawansowanego.
- HIFU (High Intensity Focused Ultrasound) to technika medyczna pozwalająca zniszczyć guz za pomocą ciepła (ablacja termiczna).
- Brachyterapia to metoda radioterapii stosowana do niszczenia komórek nowotworowych poprzez wprowadzenie do prostaty radioaktywnych implantów, które emitują promieniowanie gamma.
- Terapia hormonalna polega na przyjmowaniu analogów GnRH (gonadoliberyny), które wpływają na produkcję hormonów płciowych i mogą spowolnić postęp choroby poprzez silne obniżenie poziomu testosteronu.
- Chemioterapia jest stosowana, gdy nowotwór przestaje reagować na inne leczenie.
Powyżej 50. roku życia
Rak prostaty jest najczęstszym nowotworem złośliwym u mężczyzn powyżej 50. roku życia.
Druga przyczyna zgonów
Stanowi on drugą przyczynę zgonów z powodu raka
Rak prostaty
Po 70. roku życia
Jest diagnozowany najczęściej po 70. roku życia
Źródła :
Urologia. Ilustrowany podręcznik dla studentów i stażystów. Redakca naukowa: Tomasz Drewa, Kajetan Juszczak.PZWL Wydawnictwo Lekarskie, Warszawa 2018
Rak rdzeniasty tarczycy
Tarczyca jest gruczołem wydzielania wewnętrznego. Gruczoł ten jest zbudowany z komórek pęcherzykowych ułożonych sferycznie, które są wypełnione substancją koloidową. Hormony tarczycy (tyroksyna [T4] i 3,5,3′-trójjodotyronina [T3]) są syntetyzowane w komórkach pęcherzykowych.
T3/T4 pobudzają metabolizm wielu komórek organizmu, kontrolując częstość pracy serca, ciśnienie tętnicze krwi, temperaturę ciała i tempo podstawowych procesów metabolicznych.
Komórki C występują w otaczającej pęcherzyki tkance śródmiąższowej i wydzielają kalcytoninę. Rak rdzeniasty tarczycy (ang. medullary thyroid cancer, MTC) wywodzi się z komórek C i jest zaliczany do chorób sierocych. Może być dziedziczny (w 25% przypadków) lub sporadyczny/samoistny (w 75% przypadków).
W większości przypadków przyczyną raka rdzeniastego tarczycy jest mutacja genetyczna (zmiana w genach lub w sekwencji DNA protoonkogenu RET). Mutacja ta prowadzi do nieprawidłowej proliferacji komórek C, a w wielu przypadkach do późniejszego rozwoju raka rdzeniastego tarczycy.
Podstawową metodą leczenia raka rdzeniastego tarczycy jest operacyjne usunięcie gruczołu tarczowego w całości (tyreoidektomia całkowita).
Gdy choroba jest bardziej zaawansowana i/lub pojawiły się odległe przerzuty, pacjent może kwalifikować się do leczenia systemowego przy pomocy podawanych doustnie inhibitorów kinazy tyrozynowej.
Poniżej można znaleźć informacje na temat tej choroby, jej rozpoznania i metod leczenia.
Objawy
We wczesnym stadium zaawansowania rak tarczycy zazwyczaj nie daje żadnych objawów przedmiotowych ani podmiotowych. W miarę rozrastania się, rak tarczycy może spowodować:
- wystąpienie zgrubienia na szyi, które jest wykrywane przypadkowo lub w badaniu palpacyjnym;
- wystąpienie guzka/guzków tarczycy;
- zmiany głosu, w tym także chrypkę;
- problemy z przełykaniem;
- ból szyi lub gardła;
- powiększenie szyjnych węzłów chłonnych.
Objawy mogą być związane z miejscowym naciekaniem i zmianami przerzutowymi albo z wytwarzaniem hormonów przez nowotwór — np. biegunka, nagłe zaczerwienienie twarzy lub (rzadziej) zespół Cushinga. Nowotwór u pacjentów może być bezobjawowy, a obecność przerzutów odległych (głównie w wątrobie, płucach i kościach) wykrywa się w badaniach obrazowych, gdy stężenie kalcytoniny i antygenu rakowo-płodowego (ang. carcinoembryonic antigen, CEA) w surowicy jest podwyższone.
W wielu przypadkach rak tarczycy nie daje żadnych objawów, a chorobę wykrywa się podczas badań wykonywanych w związku z innymi dolegliwościami.
Czyinniki ryzyka
Jedynym znanym środowiskowym czynnikiem ryzyka jest narażenie na promieniowanie jonizujące. Ryzyko rozwoju raka tarczycy wywołanego przez promieniowanie jest najwyższe u dzieci.
Ponadto wyższe ryzyko zachorowania stwierdza się u kobiet oraz u pacjentów, w których rodzinie występował rak tarczycy.
Inny istotny czynnik ryzyka rozwoju raka tarczycy można przypisać zespołom dziedzicznym, takim jak mnoga gruczolakowatość wewnątrzwydzielnicza i rodzinna postać raka rdzeniastego tarczycy.
Częstość występowania (1)
Rak rdzeniasty tarczycy wywodzi się z komórek C wytwarzających kalcytoninę i stanowi 2–5% wszystkich raków tarczycy (choroba sieroca): rocznie stwierdza się 3000–4000 nowych przypadków w Europie i w Stanach Zjednoczonych. Rak rdzeniasty tarczycy może być dziedziczny (w 25% przypadków) lub sporadyczny/samoistny (w 75% przypadków). Mutację w protoonkogenie przestawionym podczas transfekcji (ang. rearranged during transfection, RET) spotyka się niemal we wszystkich przypadkach dziedzicznego raka rdzeniastego tarczycy, a mutacje somatyczne RET występują maksymalnie w 50% przypadków raka sporadycznego.
Protoonkogen RET pełni niezbędną (fizjologiczną) funkcję w komórkach C. Jednak pewne mutacje genu RET powodują, że jest on bez przerwy aktywny. Mutacja ta prowadzi do nieprawidłowej proliferacji komórek C, a w wielu przypadkach do późniejszego rozwoju raka rdzeniastego tarczycy.
Ze względu na dziedziczny charakter niektórych raków rdzeniastych tarczycy zaleca się wykonanie badań genetycznych u wszystkich pacjentów ze świeżo rozpoznanym rakiem rdzeniastym oraz u członków ich rodzin w celu wykrycia potencjalnych mutacji w protoonkogenie RET, nawet jeśli dane z wywiadu rodzinnego nie wskazują na występowanie raka rdzeniastego tarczycy.
W populacji ogólnej bardzo często występują guzki tarczycy, które można wykryć palpacyjnie.
Często ich obecność stwierdza się przypadkowo w badaniach obrazowych, takich jak badanie ultrasonograficzne (USG) szyi, obrazowanie metodą rezonansu magnetycznego lub pozytonowa tomografia emisyjna (ang. positron emission tomography, PET), wykonywanych z innych powodów.
Odsetek guzków tarczycy o potwierdzonej złośliwości wynosi od 10% do 15%.
Rak rdzeniasty tarczycy zazwyczaj wytwarza białka — kalcytoninę i antygen rakowo-płodowy (CEA) — których stężenie można oznaczyć w badaniach krwi. Białka te, w ramach długotrwałego procesu obserwacji, mogą służyć jako markery nowotworowe do wykrywania chorób nawracających lub przewlekłych.
W przypadku wszystkich podejrzanych guzków należy wykonać diagnostyczne USG tarczycy, aby potwierdzić obecność guzka i sprawdzić, czy nie ma on niepokojących cech.
Jeśli wyniki wstępnej oceny wskazują na obecność niepokojącego guzka w badaniu ultrasonograficznym, należy wykonać cienkoigłową biopsję aspiracyjną w celu potwierdzenia rozpoznania.
U większości pacjentów z rakiem rdzeniastym tarczycy rokowanie jest stosunkowo dobre, a po upływie 10 lat częstość występowania nawrotów klinicznych może wynosić od 35% do 65%. U znacznej liczby pacjentów z przerzutami odległymi choroba może mieć powolny przebieg, przy czym zmiany pozostają nieaktywne albo wolno rosną w ciągu wielu lat obserwacji.
Współczynnik 10-letniego przeżycia u pacjentów z rakiem rdzeniastym tarczycy wynosi około 75%, ale spada do 20%, gdy w momencie wstępnego rozpoznania stwierdza się chorobę w stadium przerzutów.
Podstawową metodą leczenia raka rdzeniastego tarczycy jest operacyjne usunięcie gruczołu tarczowego w całości (tyreoidektomia całkowita). Jeśli stwierdza się cechy zajęcia szyjnych węzłów chłonnych, usuwa się także węzły. Chirurgicznie usuwa się również zmiany miejscowo zaawansowane, które mogą zajmować drogi oddechowe i przewód pokarmowy. Jeśli stwierdza się masywny rozsiew nowotworu, należy starannie rozważyć korzyści i ryzyko związane z leczeniem operacyjnym. W tej sytuacji często odstępuje się od operacji.
Rak rdzeniasty tarczycy nie gromadzi jodu. Z tego względu leczenie jodem radioaktywnym jest nieskuteczne i NIE powinno być stosowane w raku rdzeniastym tarczycy.
U pacjentów z rakiem rdzeniastym tarczycy w zaawansowanym stadium nie wykazano trwałej obiektywnej odpowiedzi na radioterapię ani na chemioterapię.
Źródła :
1-Wells SA Jr, Asa SL, Dralle H, et al. American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610.
2-Davidge-Pitts CJ, Thompson GB. Thyroid tumours. In: DeVita VT, Lawrence TS, Rosenberg SA, eds. Cancer: Principles & Practice of Oncology. 10th ed. Wolters Kluwer Health. USA: Lippincott Williams and Wilkins; 2015:1-39.
3– Hadoux JU, et al. Management of advanced medullary thyroid cancer; Lancet Diabetes Endocrinol. 2016; 4:64-71.
4– Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006;107:2134–2142.
Endokrynologia
Endokrynologia jest dziedziną medycyny, która zajmuje się między innymi badaniami hormonów— substancji chemicznych, które regulują ważne funkcje ludzkiego organizmu.
Endokrynologia
Choroby endokrynologiczne, które mogą być odpowiedzialne za zaburzenia równowagi hormonalnej i metabolicznej w czasie życia pacjentów, stanowią dla medycyny ważne wyzwanie.
Akromegalia
Akromegalia, której objawy opisał po raz pierwszy Pierre Marie, jest zaburzeniem hormonalnym, które charakteryzuje się nieprawidłowym wzrostem kości oraz znacznym powiększeniem rąk i stóp, powiększeniem i pogrubieniem twarzy. Dowiedz się więcej na temat tego rzadkiego zaburzenia endokrynologicznego, sposobów jego rozpoznania oraz różnych metod leczenia.
Definicja
Akromegalia jest rzadką, przewlekłą chorobą, która prowadzi do zmian wyglądu zewnętrznego z powiększeniem twarzoczaszki, rąk i stóp, rozrostem tkanek miękkich, kości i narządów wewnętrznych oraz wielu powikłań układowych, które prowadzą do pogorszenia jakości życia i jego skrócenia.
Objawy te są spowodowane nadmiernym wydzielaniem hormonu wzrostu (GH, growth hormone) przez przysadkę mózgową, niewielki gruczoł znajdujący się w mózgu (rysunek 1). Hormon ten odgrywa ważną rolę w regulacji wzrostu dzieci i młodzieży, lecz jest również ważny u osób dorosłych. W akromegalii, pojawienie się łagodnego nowotworu (gruczolaka przysadki) wiąże się z nadmiernym wydzielaniem hormonu wzrostu.
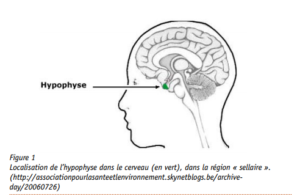
Rysunek 1
Objawy i skutki zdrowotne
Różne objawy akromegalii są albo spowodowane nadmiarem hormonu wzrostu, którego działanie na organizm jest nasilone, albo samym guzem przysadki. Objawy pojawiają się stopniowo i są widoczne dopiero po kilku latach.
Objawy związane z nadmiarem hormonu wzrostu:
- Stopniowa zmiana twarzy i kończyn (rąk i nóg): zwiększa się rozmiar buta i trudno jest zdjąć pierścionki z palców. Twarz staje się większa, charakterystyczny jest pogrubiony nos i wargi, łuki brwiowe, kości policzkowe i wystający podbródek, zwiększone odstępy między zębami oraz chrapliwy, głęboki głos.
- Zwiększenie objętości narządów wewnętrznych, głównie powiększenie wątroby, tarczycy (wole), a zwłaszcza serca, co powoduje duszność (niewydolność serca) oraz nadciśnienie tętnicze.
- Bóle pleców i stawów dotykają dwie trzecie chorych. Mogą być bardzo uciążliwe, zwłaszcza bóle palców (trudności w pisaniu, zawiązaniu butów itp.).
- Pojawiają się również odkształcenia kości, w szczególności boczne skrzywienie kręgosłupa (skolioza) lub odkształcenie mostka.
- Pojawienie się zespołu cieśni kanału nadgarstka jest bardzo powszechne, powoduje drętwienie oraz mrowienie, a następnie ból dłoni.
- Osłabienie słuchu.
- Chrapanie i bezdech senny są bardzo częste (nawet do 80% pacjentów), a towarzysząca im senność w ciągu dnia może w dłuższym okresie powodować zaburzenia pracy serca oraz choroby układu oddechowego.
- Może również pojawić się cukrzyca.
- Wyraźne zmęczenie jest także częstym objawem.
- Inne częste skutki nadmiaru hormonu wzrostu:
- przyrost masy ciała,
- pogrubienie i starzenie się skóry,
- nadmierna potliwość,
- nadmierne owłosienie.
- Małe polipy lub gruczolaki jelita grubego, które czasem mogą prowadzić do raka jelita grubego.
Objawy związane z guzem przysadki:
- Bóle głowy występują często ze względu na objętość guza przysadki.
- Zaburzenia widzenia występują ze względu na ucisk skrzyżowania nerwu wzrokowego.
- Zmniejszenie produkcji niektórych hormonów.
W związku z licznymi konsekwencjami zdrowotnymi akromegalia ma wpływ na długość życia pacjentów. Nieleczona lub rozpoznana późno choroba wiąże się ze skróceniem długości życia.
Czynniki ryzyka
Czynniki ryzyka oraz możliwości zapobiegania akromegalii nie są znane. Przed pojawieniem się objawów nie jest możliwe wykrycie tej choroby. Akromegalia nie jest dziedziczna i nie jest przekazywana dzieciom.
Częstość występowania
Akromegalia jest rzadką chorobą, notuje się około 70 przypadków na 1 milion osób. Chociaż może ona wystąpić w każdym wieku, jest często diagnozowana u osób w wieku około 40 lat i wyjątkowo wśród osób starszych.
Akromegalia jest zaburzeniem hormonalnym rozpoznawanym późno ze względu na powolne wystąpienie objawów i zmian fizycznych zachodzących podstępnie w ciągu lat. Czasami pacjent decyduje się na konsultację lekarską z powodu bólu pleców i stawów. Rozpoznanie często następuje dopiero po 5-10 latach od wystąpienia pierwszych objawów klinicznych.
W celu potwierdzenia rozpoznania akromegalii należy przeprowadzić następujące badania:
- Badanie krwi potwierdzające wysoki poziom IGF-1 (insulinopodobny czynnik wzrostu -1), obecnie uważany za najlepszy wskaźnik akromegalii.
- Test hamowania wydzielania hormonu wzrostu po podaniu glukozy, który polega na sztucznym zwiększeniu poziomu glukozy we krwi (poprzez wypicie słodkiego napoju) i na regularnym pomiarze poziomu hormonu wzrostu we krwi. U osoby zdrowej podwyższony poziom cukru we krwi powoduje zmniejszenie wydzielania hormonu wzrostu. W przypadku akromegalii poziom ten pozostaje stały i nie występuje hamowanie wydzielania tego hormonu.
- Tomografia komputerowa (CT) lub rezonans magnetyczny (MRI) mają na celu stwierdzenie obecności gruczolaka przysadki.
Podstawowym celem leczenia akromegalii jest normalizacja wydzielania GH i IGF-1. Celem drugorzędowym jest usunięcie guza lub znacząca redukcja jego objętości. Można to osiągnąć stosując leczenie operacyjne, farmakologiczne oraz radioterapię (rzadziej):
Leczenie chirurgiczne, które jest najczęstszym sposobem usunięcia gruczolaka przysadki. Zabieg jest często wykonywany przez nos, jednakże w przypadku dużych zmian konieczny jest dostęp przezczaszkowy. Operacja przywraca normalne wydzielanie hormonu wzrostu u około 70-90% pacjentów z małym gruczolakiem oraz u 30 do 50% pacjentów, u których gruczolak osiągnął średnicę powyżej 10 mm. W celu ułatwienia leczenia operacyjnego można rozważyć zastosowanie analogów somatostatyny jako przygotowania do operacji.
Jeżeli leczenie chirurgiczne nie jest możliwe albo operacja nie jest wystarczającą metodą do wyrównania poziomu krążącego IGF-1 lub hormonu wzrostu oraz w przypadku nawrotu choroby, należy zastosować inne metody leczenia, jak radioterapia lub leczenie farmakologiczne.
W przypadku niepowodzenia operacji stosuje się leczenie farmakologiczne w celu zmniejszenia wydzielania hormonu wzrostu i/lub IGF-1 przez guz. Istnieje kilka grup leków:
- analogi somatostatyny, przede wszystkim lanreotyd i oktreotyd, stosowane w celu normalizacji stężenia hormonu wzrostu, a także zmniejszenia objętości gruczolaka w ciągu pierwszych 3 miesięcy leczenia. Głównym działaniem ubocznym tych leków jest wystąpienie zaburzeń żołądkowo-jelitowych: ból brzucha, biegunka lub nadmierna zawartość tłuszczów w stolcu (biegunka tłuszczowa).
- Pasyreotyd, wprowadzony niedawno do lecznictwa, jest również analogiem somatostatyny, stosowanym w terapii drugiego rzutu po nieskutecznym leczeniu lanreotydem lub oktreotydem. Głównym jego działaniem niepożądanym jest wzrost poziomu cukru we krwi, a czasami wywołanie cukrzycy.
- Substancje pobudzające receptory dopaminergiczne, takie jak bromokryptyna i chinagolid, zmniejszają wydzielanie hormonu wzrostu przez guz. Leki te mogą powodować zaburzenia trawienia (nudności i wymioty), obniżenie ciśnienia tętniczego krwi podczas zmiany pozycji ciała z leżącej do stojącej, a także znaczne bóle głowy.
- Pegwisomant zapobiega oddziaływaniu hormonu wzrostu na jego receptor i może być stosowany pojedynczo lub w kombinacji z analogami somatostatyny, jeżeli nie są one wystarczająco skuteczne w kontrolowaniu poziomu hormonu wzrostu lub IGF-1. Jest on podawany codziennie w postaci iniekcji podskórnych. Nie wpływa korzystnie na wielkość guza.
W zależności od objawów i postępu choroby mogą być konieczne konsultacje u lekarzy różnych specjalności, jak kardiolog, diabetolog, okulista czy reumatolog.
3 — 4 nowe przypadki
Akromegalii rocznie na 1 million
40 lat
Średni wiek rozpoznania choroby
Źródło:
Bolanowski et al. Rozpoznanie i leczenie akromegalii – aktualizacja rekomendacji Polskiego Towarzystwa Endokrynologicznego 2019
Ciężki pierwotny niedobór IGF-1
Ciężki pierwotny niedobór IGF-1 (insulinopodobnego czynnika wzrostu 1 — białka pośredniczącego w działaniu hormonu wzrostu ) powoduje spowolnienie wzrostu u dzieci i młodzieży, jak również liczne zaburzenia anatomiczne, morfologiczne i fizjologiczne. Dowiedz się więcej na temat tego zaburzenia, jego diagnozowania i różnych metod leczenia.
Definicja
Ciężki pierwotny niedobór IGF-1 (lub (S)PIGFD, (Severe) Primary IGF Deficiency) jest rzadką chorobą odpowiedzialną za istotny deficyt wzrostu u dzieci i młodzieży, który stanowią zaburzenia fizyczne i psychologiczne.
IGF-1 (lub somatomedyna C) jest hormonem (białkiem złożonym z 70 aminokwasów) o strukturze zbliżonej do insuliny, produkowanym przez wątrobę. IGF-1 jest mediatorem działania hormonu wzrostu (lub GH, „growth hormone”). IGF-1 stymuluje wchłanianie glukozy, kwasów tłuszczowych i aminokwasów, umożliwiając wzrost tkanek. Hormon ten bierze również udział w rozwoju układu nerwowego i odgrywa ważną rolę w procesie nabywania i utrzymywania masy kostnej.
Pierwotny niedobór IGF-1 charakteryzuje się niskim poziomem IGF-1, bez jednoczesnego niewystarczającego wydzielania hormonu wzrostu (GH) przy braku określonej przyczyny.
Etiologia lub przyczyny
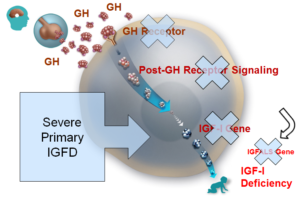
Powodem niedoboru jest nieprawidłowość kaskady GH/IGF-1 wywołującej szereg zdarzeń wewnątrzkomórkowych, która rozpoczyna się w momencie łączenia GH z receptorem (mutacje), prowadząc do wydzielania IGF-1.
Najbardziej typową formą pierwotnego niedoboru IGF-1 jest zespół Larona (opisany w roku 1999 przez Z. Larona), choroba wrodzona spowodowana mutacją genu GHR (receptora hormonu wzrostu).
Objawy i skutki zdrowotne
Skutki zdrowotne niedoboru IGF-1 w przypadku zespołu Larona sa następujące:
- bardzo niski wzrost (-2 SD* w przypadku niedoboru pierwotnego oraz -3 SD w przypadku ciężkiego niedoboru pierwotnego);
- charakterystyczne rysy twarzy, wraz z brakiem szczytu skoku pokwitaniowego;
- otyłość;
- hipogonadyzm (utrata funkcji gruczołów płciowych, zarówno w odniesieniu do produkcji hormonów, jak i wielkości gonad);
- zaburzenia metaboliczne.
Znaczenie i obecność każdego z objawów mogą być różne u różnych osób. Pozostałe objawy niedoboru pierwotnego IGF-1 występują w formie łagodniejszej niż w przypadku zespołu Larona, wraz z mniej widocznymi charakterystycznymi cechami anatomicznymi, morfologicznymi i fizjologicznymi oraz brakiem zidentyfikowanych wad genetycznych.
Częstość występowania
SPIGFD jest bardzo rzadką chorobą, częstość występowania jest mniejsza od 1 : 10 000 osób.
W praktyce pojęcie ciężkiego pierwotnego niedoboru IGF-1 może być stosowane wobec dzieci, które:
- są niskiego wzrostu (< -3 SD*),
- mają niski poziom IGF-1 w surowicy krwi (z uwzględnieniem wieku),
- mają normalny lub podwyższony poziom GH w surowicy.
*SD: odchylenie standardowe, które pozwala na dokonanie oceny stopnia rozproszenia pomiarów wokół wartości średniej.
U dzieci z pierwotnym niedoborem IGF-1 leczenie hormonem wzrostu jest nieskuteczne, ponieważ występuje u nich „blokada” kaskady GH/IGF-1.
Wśród przypadków SPIGFD wyróżnia się:
- nieprawidłowości w obrębie kaskady GH/IGF-1, które mogą być leczone rekombinowanym IGF-1,
- nieprawidłowości występujące poza kaskadą HG/IGF-1 (jak np. nieprawidłowości receptora IGF-1), które nie są wrażliwe na leczenie.
Insulinopodobny ludzki czynnik wzrostu typu 1 (IGF-1), pochodzący z rekombinowanego DNA, przeznaczony dla dzieci z ciężkim pierwotnym niedoborem IGF-1 będącym wynikiem zaburzeń kaskady GH/IGF-1, stanowi długoterminowe leczenie opóźnień wzrostu spowodowanych przez SPIGFD. Jest to lek stosowany u dzieci przez lekarza specjalistę po uwzględnieniu przeciwwskazań. Skuteczność leczenia, podawanego w formie iniekcji podskórnych dwa razy na dobę, należy oceniać na podstawie tempa wzrostu dziecka. Dawki są stopniowo zwiększane, jeżeli leczenie jest dobrze tolerowane przez pacjenta.
Źródła:
1. Cohen J. et al. Managing the Child with Severe Primary Insulin-Like Growth Factor-1 Deficiency (IGFD); 2014 Drugs R D
2. Sultan M., Afzal M., Oureshi SM.,et al.: Etiology of short stature in children. J Coll Physicians Surg Pak., 2008: (8),493-497
3. Charakterystyka Produktu Leczniczego Increlex
Lokalne badania kliniczne
Odkryj nasze badania kliniczne
BADANIE LEKU DYSPORT U PACJENTÓW ZE SPASTYCZNOŚCIĄ KOŃCZYNY GÓRNEJ PO UDARZE MÓZGU
Ocena efektywności leku Dysport u pacjentów ze spastycznością kończyny górnej po udarze mózgu
Badanie obserwacyjne na terenie Rzeczpospolitej Polskiej
Zgoda Komisji Etycznej 24 02 2015 nr 110/2015/KB/VI
Rejestracja na stronie clinicaltrials.gov ID: A-38-52120-215
Liczba ośrodków badawczych: 7
Liczba pacjentów: 108
Badanie zostało zakończone i jest analizowane.
Neurologia
Neurologia zajmuje się badaniami ośrodkowego układu nerwowego, którego uszkodzenia mogą być przyczyną ciężkich zaburzeń motorycznych niekorzystnie wpływających na jakość życia oraz samodzielność pacjentów.
Neurologia
Neurologia zajmuje się badaniami ośrodkowego układu nerwowego, którego uszkodzenia mogą być przyczyną ciężkich zaburzeń motorycznych niekorzystnie wpływających na jakość życia oraz samodzielność pacjentów.
Blefarospazm
Kurcz powiek jest chorobą, której objawem są powtarzające się mimowolne skurcze mięśni powiek. Zaburzenie to może również objawiać się przyspieszonym mruganiem, jak również całkowitą niezdolnością do otwarcia powieki, co zmniejsza zdolność pacjenta do wykonywania codziennych czynności. Dowiedz się więcej na temat tej choroby, jej diagnozowania i różnych metod leczenia.
Definicja
Kurcz powiek jest drugą najczęstszą postacią dystonii ogniskowych u dorosłych. Powoduje skurcze mięśni powiek, co z kolei prowadzi do ich niekontrolowanego zamykania. Skurcze mięśni mogą stać się praktycznie ciągłe i powodować wrażenie ślepoty u chorych, nawet jeśli ich oczy są zdrowe. Mimo, że kurcz powiek dotyczy czasami tylko jednej strony twarzy, najczęściej zaburzenia dotyczą dwóch powiek jednocześnie.
Objawy i skutki zdrowotne
Często kurcz powiek pojawia się stopniowo, a mimowolne zamykanie powiek zazwyczaj rozpoczyna się od pojedynczych mrugnięć. W większości przypadków dyskomfort jest odczuwalny podczas wzmożonej aktywności wzrokowej (czytanie, patrzenie na ekran, prowadzenie pojazdu). Po kilku tygodniach skurcze powodują całkowite zamknięcie powiek na kilka sekund lub nawet minut. Kurcz powiek może prowadzić nawet do ślepoty czynnościowej, co stanowi znaczne utrudnienie codziennego życia pacjentów.
Choroba ta może być związana ze skurczem mięśni dolnej części twarzy – zespół Meige’a, który polega na współwystępowaniu kurczu powiek i dystonii ustno-żuchwowej.
Częstotliwość występowania skurczów zazwyczaj zmienia się w ciągu dnia. W godzinach porannych są one zazwyczaj dosyć odległe w czasie, natomiast ich częstość wzrasta w godzinach popołudniowych. Pewne sytuacje mogą nasilać występowanie tego zaburzenia:
- narażenie na zbyt silne oświetlenie,
- prowadzenie samochodu,
- oglądanie telewizji,
- czytanie,
- stres lub przemęczenie.
Noszenie specjalnych okularów lub odpoczynek mogą zmniejszać objawy kurczu powiek.
Czynniki ryzyka
Przyczyna kurczu powiek, jak i zespołu Meige’a, nie jest jeszcze znana. Dlatego też mówi się o samoistnym lub idiopatycznym kurczu powiek. Należy jednakże zidentyfikować zwiększone ryzyko rozwoju choroby u osób o dużej wrażliwości na światło, cierpiących na zespół suchego oka lub przyjmujących niektóre leki, np. stosowane w leczeniu choroby Parkinsona oraz należące do grupy benzodiazepin.
Częstość występowania
Kurcz powiek jest rzadką chorobą, w Europie występuje u około 36 osób na 1 milion. Jednak liczba ta jest prawdopodobnie niedoszacowana ze względu na trudności w diagnostyce tej choroby. Najczęściej kurcz powiek pojawia się między 50 a 60 rokiem życia i występuje częściej u kobiet niż mężczyzn (średnio trzy kobiety na dwóch mężczyzn).
Diagnoza stawiana jest najczęściej przez lekarza rodzinnego na postawie wywiadu i opisu objawów choroby przez pacjenta. Do tej pory nie istnieje żadne badanie diagnostyczne, które mogłoby potwierdzić diagnozę.
Typowe objawy kurczu powiek występują również w przypadku innych zaburzeń, nie należy go mylić z:
- ptozą: opadnięciem powieki, które jest powodowane przez osłabienie lub porażenie mięśnia dźwigacza powieki górnej;
- połowiczym kurczem twarzy, który jest niedystonicznym, mimowolnym skurczem mięśni jednej strony twarzy, spowodowanym uszkodzeniem nerwu twarzowego w miejscu jego wyjścia z mózgu. Zaburzenie to często powoduje wyraźniejsze mruganie oka.
Objawy kurczu powiek mogą być zmniejszone lub kontrolowane poprzez zastosowanie jednej z następujących metod:
- Iniekcje toksyny botulinowej typu A
- Produkty lecznicze skuteczne w kurczu powiek:
- leki zwiotczające mięśnie, które rozluźniają mięśnie i łagodzą skurcz;
- leki przeciwcholinergiczne, które ograniczają działanie acetylocholiny, neuroprzekaźnika w układzie nerwowym;
- benzodiazepiny, które zmniejszają niepokój i łagodzą bolesne skurcze.
- Leczenie chirurgiczne — stosowane w najcięższych formach skurczu powiek po niepowodzeniu terapii toksyną botulinową. Po konsultacji z lekarzem okulistą możliwe jest dokonanie resekcji mięśni okrężnych powiek, zarówno samych, jak i wraz z mięśniami czoła, mięśni marszczących brwi (zlokalizowanych wzdłuż łuku brwiowego) lub mięśni znajdujących się między brwiami.
Choroba
rzadka
50 a 60 lat
to wiek, w jakim zazwyczaj pojawia się choroba”
Kobiety
Częściej występuje u kobiet niż u mężczyzn
Źródło:
„Toksyna botulinowa w praktyce neurologicznej” pod redakcją Jarosława Sławka i Moniki Rudzińskiej, tom 1. Via Medica, Gdańsk 2015
Dystonia szyjna
Dystonia szyjna, zwana również kręczem karku (spasmodic torticollis), objawia się okresowymi lub ciągłymi skurczami mięśni szyi, a czasami ramion, powodującymi nieprawidłowe ułożenie lub ruchy skręcające głowy i szyi. To zaburzenie neurologiczne pojawia się stopniowo i mogą mu towarzyszyć drżenia głowy, bóle szyi oraz uniesienie barków.
Dowiedz się więcej na temat tego zaburzenia, sposobów jego diagnozowania i różnych metod leczenia.
Definicja
Dystonia szyjna (lub kurczowy kręcz szyi) jest najbardziej rozpowszechnioną formą dystonii ogniskowej. Dystonia jest wynikiem zaburzeń neurologicznych, przejawia się skurczami mięśni, które powodują nieprawidłowe ruchy szyi, głowy i czasem ramion. Mięśnie szyi mogą się kurczyć, obracać, drżeć lub zostać zablokowane w jednej pozycji, co skutkuje nieprawidłową i niewygodną postawą.
Objawy i skutki zdrowotne
Objawy dystonii szyjnej zwykle pojawiają się stopniowo: głowa przyjmuje nieprawidłową postawę co jakiś czas — nie powoduje to dyskomfortu i często na początku jest zauważane jedynie przez otoczenie, a nie przez samego chorego. Choroba może ujawnić się pod wpływem innej choroby w czasie jej wystąpienia. Dystonia przejawia się:
- skurczami krótkimi lub przedłużonymi, które powodują nieprawidłową i utrwaloną postawę głowy;
- drżeniami głowy;
- bólami szyi;
- uniesieniem barków.
Z czasem dystonia szyjna powoduje mimowolne odchylenie głowy, które może przybierać różne formy:
- torticollis: kręcz rotacyjny szyi w prawo lub w lewo (wystepuje u ponad 50% chorych);
- laterocollis: przechylenie szyi w kierunku ramienia (od 40 do 70% przypadków);
- retrocollis: zgięcie szyi ku tyłowi;
- anterocollis: przechylenie szyi do przodu (najrzadsza forma);
A także:
- laterocaput: pochylenie głowy w kierunku ramienia
- torticaput: skręt głowy w kierunku ramienia
- anterocaput: pochylenie głowy do przodu
- retrocaput: pochylenie głowy do tyłu
Często obserwuje się mieszaną formę dystonii szyjnej, na którą składają się elementy kilku wymienionych powyżej ruchów. Przebieg choroby jest zmienny. W ciągu pierwszych pięciu lat choroby w około 5% do 20% przypadków nastepuje remisja, która może trwać od kilku miesięcy do kilku lat, po czym może nastąpić nawrót objawów. W większości przypadków objawy nasilają się w ciągu pierwszych pięciu lat, a następnie ulegają stabilizacji. Inni pacjenci doświadczają następujących po sobie okresów zaostrzania oraz wyciszania choroby. W rzadkich przypadkach dystonia może rozprzestrzeniać się na sąsiadujące części ciała.
Czynniki ryzyka
Przyczyny dystonii szyjnej nie są jeszcze dobrze znane. Dystonia szyjna może być pierwotna lub wtórna. Dystonia pierwotna jest przeważnie chorobą idiopatyczną (o nieznanym pochodzeniu), jednak czynniki genetyczne zdają się odgrywać ważną rolę (np. typy dystonii DYT1, DYT6, DYT25 z mutacjami w genach – odpowiednio: TOR1A, THAP1, GNAL). Dystonia wtórna występuje z powodu zaburzeń metabolicznych lub zaburzeń danej struktury, zwykle w połączeniu z innymi zaburzeniami neurologicznymi. Przyczynami dystonii wtórnej mogą być zaburzenia metaboliczne lub strukturalne uszkodzenia powstałe w wyniku udaru, urazu, procesów rozrostowych bądź demielinizacyjnych lub zapalnych, zwykle w połączeniu z innymi zaburzeniami neurologicznymi.
Częstość występowania
Dystonia szyjna jest najbardziej rozpowszechnioną formą dystonii ogniskowych.
W Europie występuje u około 57 osób na 1 milion. Kurczowy kręcz szyi pojawia się zwykle około 40 roku życia i u większości pacjentów rozwija się między 30 a 60 rokiem życia. Dystonia może jednak występować na wcześniejszym etapie życia, w dzieciństwie lub u młodych dorosłych. Dystonia szyjna dotyka nieco częściej kobiety niż mężczyzn.
Jeżeli odchylenia głowy są sporadyczne, dystonia szyjna może pozostać nierozpoznana przez wiele lat. Diagnoza zwykle stawiana przez lekarza ogólnego lub neurologa opiera się na stwierdzeniu:
- ograniczonego ruchu szyi pacjenta;
- nieprawidłowego położenia głowy i/lub szyi, z drżeniem lub bez;
- zauważalnego zgrubienia niektórych lub wszystkich dotkniętych mięśni.
Możliwe jest również wykonanie badania elektromiograficznego (EMG) w celu dokonania pomiaru aktywności mięśni w gabinecie lekarskim lub na szpitalnym oddziale neurofizjologii. W przeciwieństwie do zwykłego badania EMG wykonywanego przy użyciu cienkiej igły wkłuwanej do mięśnia, badanie odbywa się za pomocą elektrod przyklejanych do skóry w miejscu, w którym mają być rejestrowane nieprawidłowe ruchy mięśni charakterystyczne dla dystonii.
W zależności od nasilenia dystonii szyjnej rozważa się różne metody leczenia:
- Iniekcje toksyny botulinowejtypu A — neurotoksyna blokuje impulsy nerwowe między nerwem a mięśniem, co powoduje ograniczenie skurczy mięśni na najczęściej około trzy miesiące.
- Leczenie farmakologiczne — istnieje kilka grup leków, które mogą oddziaływać na objawy dystonii szyjnej:
- leki zwiotczające mięśnie, które rozluźniają mięśnie i łagodzą skurcze;
- leki przeciwcholinergiczne, które ograniczają działanie acetylocholiny, neuroprzekaźnika w układzie nerwowym;
- benzodiazepiny, które zmniejszają niepokój i łagodzą bolesne skurcze;
- środki przeciwbólowe.
- Zabieg chirurgiczny — stosowany w przypadku najcięższych form dystonii po niepowodzeniu innych metod terapeutycznych oraz po dokonaniu oceny neurofizjologicznej. W przypadku dystonii szyjnej preferowane jest stosowanie selektywnej oraz obwodowej techniki neurochirurgicznej (poza kręgosłupem).
- Fizjoterapia — uzupełnia iniekcje z toksyny botulinowej lub farmakoterapię. Specjalnie dobrane ćwiczenia pomagają przywrócić równowagę między mięśniami, które są nadmiernie lub niewystarczająco pobudzone, tak aby pacjent mógł odzyskać naturalną oś i amplitudę obrotu głowy. Aby fizjoterapia była skuteczna, musi być wspomagana ćwiczeniami rehabilitacyjnymi codziennie wykonywanymi samodzielnie przez pacjenta.
Najbardziej rozpowszechniona
Forma dystonii ogniskowych
Między 30 a 60 rokiem życia
pojawia się zazwyczaj choroba
Zaburzenie neurologiczne
układu ruchu o stopniowym rozwoju
Źródło:
„Toksyna botulinowa w praktyce neurologicznej” pod redakcją Jarosława Sławka i Moniki Rudzińskiej, tom 1. Via Medica, Gdańsk 2015
Nadmierna potliwość
Nadmierne pocenie się, czyli nadpotliwość, oznacza nadmierną produkcję potu przekraczającą ilość potrzebną do termoregulacji organizmu. Obszarami najczęściej dotkniętymi tym zaburzeniem są pachy, dłonie, stopy i twarz, ale może ono również występować na całej powierzchni ciała. Dowiedz się więcej na temat tej choroby, jej rozpoznawania i różnych metod leczenia.
Definicja
Nadpotliwość oznacza nadmierną produkcję potu, która może być przerywana lub stała, i nasilać się podczas sytuacji kryzysowych.
Istnieją różne odmiany nadpotliwości:
- Nadpotliwość ogólna: często występuje z powodu przewlekłego zakażenia, choroby metabolicznej lub chłoniaka i powoduje pocenie się na całej powierzchni ciała.
- Nadpotliwość miejscowa pierwotna: idiopatyczna (bez rozpoznanej przyczyny) to nadmierne pocenie się przez ponad sześć miesięcy ograniczone do pach, stóp, pachwin i głowy.
- Nadpotliwość miejscowa wtórna lub strefowa: występuje często z powodu zaburzeń neurologicznych, jest ograniczona do połowy ciała lub do kończyny.
Objawy i skutki zdrowotne
Nasilenie objawów jest bardzo zróżnicowane. Wyróżnia się cztery typy nadmiernej potliwości: [1]
- Poziom 1: Pocenie niewidoczne, nie wpływa na codzienną aktywność
- Poziom 2: Pocenie w zakresie tolerancji, tylko czasem zaburza codzienną aktywność
- Poziom 3: Pocenie na granicy tolerancji, często zaburza codzienną aktywność
- Poziom 4: Pocenie nietolerowane, stale zaburza codzienną aktywność
W najcięższych przypadkach nadmierna potliwość powoduje:
- zaczerwienienie (rumień);
- obrzęki;
- bóle;
- znaczne ochłodzenie ciała i kończyn (z powodu parowania);
- niedogodności towarzyskie związane z nadmiernym poceniem się, co może prowadzić do niepokoju, tachykardii oraz nadmiernego unaczynienia.
Nadmierne pocenie się jest często jeszcze bardziej wzmożone w sytuacjach stresowych i podczas silnych emocji, w cieple, w czasie gorączki lub spożywania niektórych pokarmów (takich jak kawa, czekolada i przyprawy).
Czynniki ryzyka
Przyczyna choroby jest często nieznana, jest ona wynikiem nadmiernej aktywacji odruchów nerwowych odpowiedzialnych za pocenie się. Nadmierna potliwość pod pachami najczęściej pojawia się bez widocznej przyczyny, zazwyczaj przed 25 rokiem życia. Wskazuje się jednak czynnik genetyczny, gdyż w wywiadzie rodzinnym występuje ona w 42% przypadków.
Częstość występowania
Nadmierne pocenie się jest rzadką chorobą, która występuje u około 3% populacji w Stanach Zjednoczonych i Niemczech, a jedynie 1% w Wielkiej Brytanii. Liczba chorych we Francji pozostaje nieznana.
Choroba ta występuje zwykle w okresie dojrzewania lub u młodych dorosłych (w szczególności w przypadku nadmiernego pocenia się pod pachami), osiąga swój szczyt między 30 a 40 rokiem życia, a następnie zmniejsza się stopniowo wraz z wiekiem. Odnotowuje się także kilka przypadków nadpotliwości dłoni u dzieci.
Na początek należy ocenić nasilenie pocenia. W tym celu ocenia się ilość potu (chociaż może być zmienna w czasie) za pomocą testu Minora. Test polega na naniesieniu na dany obszar skóry roztworu jodyny, a następnie posypaniu skrobią ziemniaczaną. Obszary, w których pocenie się jest nadmierne, ciemnieją, zaś obszary o mniejszej potliwości pozostają w kolorze jasnobrązowym. Należy odpowiednio ocenić psychologiczne i społeczne skutki nadmiernego pocenia i dobrać skuteczne środki lecznicze.
Istnieje wiele sposobów przydatnych w kontroli nadmiernego pocenia się, które stosuje się zgodnie z poniższą kolejnością (od leczenia miejscowego po leczenie chirurgiczne), zaczynając od najmniej agresywnych sposobów:
- Antyperspiranty miejscowe: antyperspiranty, takie jak sole glinu (najczęściej) wykazują dużą skuteczność przeciwko wydzielaniu potu, szczególnie w przypadku nadmiernego pocenia się pod pachami. Antyperspiranty tworzą czop zatykający przewody potowe, a następnie inicjują reakcję chemiczną, która absorbuje wodę, zmniejszając miejscową wilgotność. Kwasowość wytwarzana w wyniku reakcji pozwala również zmniejszyć florę bakteryjną i grzybiczą, ale jest odpowiedzialna za podrażnienie skóry (które może być zminimalizowane poprzez zastosowanie chlorku glinowego na suchą skórę).
- Jonoforeza: Poprzez zanurzenie rąk pacjenta w wodzie, przez którą przepływa prąd o stałym natężeniu, jonoforeza pomaga w stabilizacji komórek gruczołów potowych. Zabieg ten, wykonywany w niektórych gabinetach dermatologicznych oraz szpitalach powinien być wykonywany 2–3 razy w tygodniu przez miesiąc, a następnie raz w miesiącu, aby utrzymać odpowiednie wyniki.
- Iniekcje toksyny botulinowej typu A: zabieg ten jest bardzo skuteczny w przypadku nadmiernej potliwości i szybko poprawia jakość życia pacjentów. Dzięki iniekcji do skóry właściwej toksyna botulinowa hamuje skurcze komórek mięśniowych o właściwościach kurczliwych znajdujących się wokół gruczołów potowych i zapobiega ich opróżnianiu. Obszary dotknięte nadmiernym poceniem są identyfikowane na podstawie testu Minora, a następnie wstrzykuje się do nich toksynę wielopunktowo, w odległości co 1 cm (rys. 3). Zastrzyki pod pachami są niebolesne. Ich skuteczność utrzymuje się od 4 do 25 miesięcy.
- Leczenie chirurgiczne: Istnieją dwa rodzaje procedur chirurgicznych, które są zarezerwowane dla obfitych form pocenia się po niepowodzeniu zastosowania toksyny botulinowej. Sympatektomia piersiowa polega na resekcji odcinka nerwu współczulnego na poziomie klatki piersiowej, co powoduje całkowite zatrzymanie pocenia się w górnej części ciała. Może to jednak powodować uciążliwe skutki uboczne, takie jak nadmierne pocenie wyrównawcze (pocenie pozostałych obszarów po zabiegu), które może być bardzo niekomfortowe, jeszcze bardziej niż nadpotliwość początkowa. W przypadku nadmiernego pocenia się pod pachami można również usunąć znajdujące się tam gruczoły potowe. Powikłania takiej operacji są rzadkie, ale u pacjentów pozostaje duża blizna na całe życie.
4
Poziomy nasilenia
Choroba
rzadka
Połowiczy kurcz twarzy
Połowiczy kurcz twarzy (HFS, Hemifacial Spasm) charakteryzuje się jednostronnymi, mimowolnymi skurczami mięśni, unerwianymi przez nerw twarzowy. To zaburzenie mięśniowe może w końcu stać się przewlekłe i spowodować ciężką niepełnosprawność społeczną. Dowiedz się więcej na temat tej choroby, jej diagnozowania i różnych metod leczenia.
Definicja
Mimo wielu podobieństw, połowiczy kurcz twarzy nie jest dystonią. Charakteryzuje się mimowolnymi, jednostronnymi skurczami twarzy w regionach unerwionych przez nerw twarzowy (czoło, brwi, powieki, spoidło wargi). Zwykle zaczyna się okazjonalnymi skurczami powieki, a następnie rozprzestrzenia się na inne mięśnie po tej samej stronie twarzy, jak również na powierzchowne mięśnie szyi.
Objawy i skutki zdrowotne
Choroba pojawia się stopniowo, początkowo występują skurcze powieki, a następnie może rozprzestrzeniać się na inne obszary po tej samej stronie twarzy (mięśnie policzkowe, wargi i podbródek) oraz szyi. W połowiczym kurczu twarzy może występować charakterystyczny, jednostronny grymas na twarzy chorego, nierzadko z silnym mruganiem oka, rozciągnięciem ust, skurczem czoła oraz uniesieniem jednej brwi powyżej oczodołu (drugi objaw Babińskiego). Niektórzy pacjenci słyszą specyficzny odgłos ze względu na skurcz małego mięśnia ucha podczas kurczu.
Te niekontrolowane skurcze mięśni są początkowo bardzo krótkie i rzadkie. Mimo, że często obserwowane są okresy remisji, z upływem czasu skurcze zwykle się nasilają i trwają coraz dłużej, tworząc w zasadzie stałe odkształcenie twarzy, co może prowadzić do ślepoty czynnościowej jednego oka. W życiu codziennym objawy te mogą być wzmożone w stanach zmęczenia lub stresu i zwykle ustępują w czasie snu.
Choroba ta znacznie zakłóca codzienne funkcjonowanie, zarówno jeśli chodzi o wzrok, jak i zakłóca relacje społeczne ze względu na wykrzywienie grymasu twarzy.
Czynniki ryzyka
Połowiczy kurcz twarzy może być spowodowany uszkodzeniem nerwu twarzowego poprzez jego ucisk (przez naczynie lub guz) lub uszkodzeniami innymi niż kompresyjne (chorobami neurologicznymi, urazami). O ile przyczyny te są powszechne, często zdarza się, że lekarze nie są w stanie wykryć czynnika wyzwalającego chorobę.
Częstość występowania
Połowiczy kurcz twarzy występuje zwykle między 50 a 70 rokiem życia i dotyka częściej kobiety niż mężczyzn.
Rozpoznanie połowiczego kurczu twarzy opiera się na badaniach, w trakcie których nie potwierdzą się inne objawy niż skurcze mięśni unerwianych przez mięsień twarzowy.
- Wywiad oraz zbadanie pacjenta przez lekarza ogólnego, którego zadaniem jest rozróżnienie połowiczego kurczu twarzy od innych, podobnych zaburzeń ruchowych, takich jak kurcz powiek, tiki twarzy, miokimie (powolne fale skurczu na całej długości mięśnia) lub kurcz twarzy po porażeniu.
- Badanie MRI mózgu (badanie metodą rezonansu magnetycznego), które wykonuje się dosyć często w celu postawienia diagnozy. Badanie to, połączone z angiografią MR (badanie MRI naczyń krwionośnych) pomaga także wykryć konflikt nerwowo-naczyniowy (ucisk tętnicy na nerw twarzowy), który występuje u nawet do 96% pacjentów. W tym przypadku należy rozważyć przeprowadzenie zabiegu neurochirurgicznego.
Istnieje kilka metod leczenia pacjentów z połowiczym kurczem twarzy:
- Iniekcje toksyny botulinowej typu A wykonywane przez lekarza specjalistę (neurologa). Toksyna jest wstrzykiwana do mięśnia okrężnego oka (mięsień tworzący obszar eliptyczny wokół powiek), a czasami do mięśni dolnej części twarzy. Neurotoksyna botulinowa może ograniczyć sygnał nerwowy dochodzący do mięśni i zmniejszyć ich skurcze. Efekty tego zabiegu utrzymują się przez około trzy miesiące. Zabieg może być powtarzany wielokrotnie, z zachowaniem odpowiednich odstępów czasowych.
- Zabieg chirurgiczny stosowany jako leczenie alternatywne wobec toksyny botulinowej, zwłaszcza w przypadku wtórnego połowiczego kurczu twarzy, spowodowanego uszkodzeniem. Operacja w tym przypadku jest często najlepszą metodą leczenia u młodszych pacjentów. Zabieg polega na odizolowaniu nerwu twarzowego od uciskającej go tkanki (tętnicy bądź guza).
- Różnego rodzaju środki farmakologiczne stosowane w leczeniu połowiczego kurczu twarzy, takie jak środki przeciwpadaczkowe i przeciwcholinergiczne, neuroleptyki. Wykazują one jednak względną skuteczność działania.
Choroba zazwyczaj pojawia się między 50 a 70 rokiem życia
Częściej występuje u kobiet
MRI mózgu umożliwia postawienie dokładnej diagnozy
Źródło:
„Toksyna botulinowa w praktyce neurologicznej” pod redakcją Jarosława Sławka i Moniki Rudzińskiej, tom 1. Via Medica, Gdańsk 2015
Porażenie mózgowe
Dziecięce porażenie mózgowe odnosi się do różnych niepełnosprawności ruchowych wynikających z uszkodzenia mózgu w czasie ciąży, podczas porodu lub w okresie niemowlęctwa. Zaburzeniom ruchowym często towarzyszą zaburzenia funkcji poznawczych, opóźnienie rozwoju umysłowego oraz ból, które znacznie wpływają na jakość życia i samodzielność pacjenta. Dowiedz się więcej na temat tej choroby, jej diagnozowania i różnych metod leczenia.
Definicja
Pojęcie porażenia mózgowego obejmuje różne trwałe zaburzenia rozwojowe ruchu i postawy, odpowiedzialne za ograniczenie aktywności, spowodowane niepostępującymi uszkodzeniami powstałymi podczas rozwoju mózgu u płodu lub noworodka na poziomie neuronów ruchowych. Zaburzeniom ruchowym porażenia mózgowego często towarzyszą zaburzenia czuciowe, percepcyjne, poznawcze, a także problemy z komunikacją i zachowaniem, padaczka oraz wtórne problemy układu mięśniowo-szkieletowego.
Termin „porażenie mózgowe” jest używany w kontekście międzynarodowym jako określenie wtórnych, niepostępujących objawów uszkodzenia mózgu.
Objawy i skutki zdrowotne
Porażenie mózgowe objawia się w różny sposób u każdego pacjenta i może łączyć zaburzenia ruchowe, czuciowe i intelektualne. Objawy, zwykle obserwowane u dzieci od szóstego miesiąca życia, obejmują:
- spastyczność (nadmierna sztywność mięśni z powodu mimowolnego zwiększonego napięcia), która, jeżeli obejmuje nogi, cechuje się wpół zgiętą pozycją, chodem krzyżowym z wyciągniętymi stopami oraz brakiem równowagi; spastyczności często towarzyszy ból;
- nietypowe położenie nóg (pozycja siedząca w kształcie litery W) oraz przewaga ruchów jednej strony ciała;
- hipotonię mięśniową (nieprawidłowe trzymanie głowy oraz trudności w utrzymaniu pozycji siedzącej);
- zmniejszone umiejętności manualne (trudności w jedzeniu, ubieraniu się, pisaniu oraz chwytaniu przedmiotów);
- trudności w połykaniu i mówieniu (dyspraksja oralna);
- esotropię (zez zbieżny);
- zanik mięśni, powolny lub niesymetryczny wzrost;
- nadwrażliwość na hałas lub przeciwnie, zmniejszoną wrażliwość słuchową;
- nadmierne zmęczenie;
- niedojrzałość emocjonalną i nadmierne reakcje w różnych sytuacjach;
- widoczny stopień upośledzenia umysłowego u niektórych pacjentów.
Jeżeli uszkodzenie mózgu nie jest postępujące, wzrost dziecka oraz obecność spastyczności mogą powodować inne objawy pojawiające się w czasie:
- deformacje kości (często na poziomie ortopedycznym);
- problemy stawowe ze względu na skurcze mięśni;
- nieprawidłowa postawa (spastyczność przywodzicieli powoduje wysokie ryzyko obciążenia stawu biodrowego).
Etiologia porażenia mózgowego
Chociaż najczęstszymi przyczynami porażenia mózgowego są niedotlenienie i niedokrwienie (zmniejszenie lub nawet zatrzymanie dopływu krwi do pewnych części mózgu, co powoduje niedobór tlenu) lub krwotok mózgowy, powodem tych zmian może być wiele innych czynników, zarówno przed porodem, w momencie narodzin, jak i u dzieci zazwyczaj poniżej drugiego roku życia:
- Przed porodem komórki mózgowe płodu mogą zostać zniszczone w wyniku udaru mózgu, wady ośrodkowego układu nerwowego lub nieprawidłowości łożyska oraz pępowiny. Zatrucie matki w wyniku przyjmowania niektórych leków lub narkotyków lub zakażenie wirusami, takimi jak wirus różyczki, toksoplazmozy i cytomegalii, mogą być również odpowiedzialne za nieodwracalne uszkodzenia mózgu u płodu. W przypadku prenatalnego porażenia mózgowego uszkodzenie pojawia się często w pierwszych miesiącach ciąży, a przyczyna zwykle pozostaje nieznana. Bardzo mała waga urodzeniowa i wcześniactwo stanowią również zwiększone ryzyko wystąpienia porażenia mózgowego.
- W przypadku terminowego rozwiązania ciąży, trudny poród oraz nieprawidłowa pozycja pępowiny mogą odciąć dopływ krwi do mózgu i spowodować porażenie mózgowe u dziecka.
- Po urodzeniu silne drgawki, wypadek, zatrzymanie pracy serca lub zakażenie (takie jak zapalenie opon mózgowych lub zapalenie mózgu) są czynnikami sprzyjającymi wystąpieniu porażenia mózgowego.
Częstość występowania
Porażenie mózgowe występuje u 1 do 3 dzieci na 1000 żywych urodzeń w krajach rozwiniętych.
Jeżeli porażenie mózgowe wystąpi w czasie ciąży lub podczas porodu, w celu rozpoznania choroby należy zwykle poczekać, aż dziecko osiągnie wiek kilku miesięcy. Diagnoza jest zwykle stawiana, gdy dziecko ma od 3 do 18 miesięcy, kiedy rodzice stwierdzają opóźnienie w rozwoju.
Rozpoznanie porażenia mózgowego opiera się na kilku badaniach:
- silna dominacja jednej kończyny w stosunku do drugiej — fakt, że dziecko zawsze chwyta przedmioty tą samą ręką, jest objawem mogącym sugerować porażenie mózgowe;
- utrzymywanie się odruchów pierwotnych (odruchy specyficzne dla noworodków, np. odruch Moro) jest możliwym objawem tej choroby;
- badanie obrazowe metodą rezonansu magnetycznego (MRI) lub echotomografii umożliwiające obejrzenie połączeń tkanek miękkich, kości i naczyń krwionośnych może wykazać fizyczne uszkodzenie mózgu;
- testy inteligencji oraz badania wzroku i słuchu pozwalają uzupełnić diagnozę przez rozpoznanie innych problemów związanych z porażeniem mózgowym.
Istnieje kilka metod leczenia spastyczności u pacjentów z porażeniem mózgowym:
- Iniekcje toksyny botulinowej typu A zmniejszają spastyczność kończyn dotkniętych chorobą.
Neurotoksyna botulinowa ogranicza sygnał nerwowy dochodzący do mięśni i zmniejsza ich skurcze. Efekty tego zabiegu utrzymują się przez około trzy miesiące i dłużej. Po tym czasie zabieg może zostać powtórzony. - Leczenie chirurgiczne: jeżeli iniekcje toksyny botulinowej nie są wystarczająco skuteczne wobec spastyczności, zabieg operacyjny może pozwolić wydłużyć skurczone mięśnie i poprawić ich ruchliwość.
- Leczenie farmakologiczne: leki zwiotczające mięśnie, takie jak baklofen i diazepam, wykazują skuteczne działanie w spastyczności. W przypadku wystąpienia drgawek wskazane jest stosowanie leków przeciwdrgawkowych.
- Leczenie farmakologiczne lub chirurgiczne może uzupełniać fizjoterapia w celu zmniejszenia spastyczności. Ćwiczenia rozciągające pozwalają utrzymać większą autonomię ruchu stawów i zapobiegają powstawaniu przykurczy mięśniowych oraz czasami bolesnej sztywności mięśni.
- Zastosowanie szyn oraz ortez zalecanych przez ortopedę, neurologa lub fizjoterapeutę ma na celu poprawę postawy i ułatwienie chodzenia.
1 do 3
Na 1000 żywych urodzeń w krajach uprzemysłowionych
Od 3 do 18 miesięcy
to wiek, w jakim zwykle rozpoznaje się porażenie mózgowe
Stopa końska
Stopa końska lub spastyczna stopa końsko-szpotawa to zniekształcenie stopy, zazwyczaj pojawiające się w wyniku udaru mózgu lub porażenia mózgowego, które może również stanowić wadę wrodzoną. Choroba upośledza chód pacjentów i stanowi dużą niepełnosprawność w zakresie motoryki. Dowiedz się więcej na temat tej choroby, jej diagnozowania i różnych metod leczenia.
Definicja
Stopa końska to deformacja stopy polegająca na uniesieniu pięty ku górze. Chodzenie odbywa się na palcach.
Końskie ustawienie stopy jest spowodowane następującymi czynnikami:
- niekontrolowanymi skurczami mięśni (spastycznością) łydki w następstwie uszkodzenia mózgu, które mogą wywoływać niezdolność do wykonywania zgięcia grzbietowego (ruchu polegającego na podniesieniu stopy w kierunku kości piszczelowej) stopy i palców;
- wadą jednej lub często dwóch stóp pojawiającą się w życiu płodowym. Jest ona przyczyną wrodzonej dysplazji (wady lub zniekształcenia wynikających z anomalii rozwojowych) wszystkich tkanek (kości, więzadeł, nerwów, naczyń krwionośnych) znajdujących się poniżej kolana.
Objawy i skutki zdrowotne
Zniekształcenie typu stopa końska może być mniej lub bardziej poważne. Nazywa się ją dynamiczną, jeżeli rozciąganie mięśni łydki jest nadal możliwe. Rozciąganie może być utrudnione poprzez zwłóknienie (zmianę włóknistą powstałą w następstwie znacznego zniszczenia tkanki) — mówi się wtedy o statycznej stopie końskiej.
Zaburzenia chodu (na przedniej części stopy) spowodowane stopą końską mają wpływ na mobilność i równowagę oraz charakteryzują się nieprawidłowym ustawieniem kostki. Utykanie, jakie powoduje końskostopie, wpływa również negatywnie na samopoczucie psychiczne oraz życie społeczne pacjentów.
Etiologia
Główną przyczyną stopy końskiej u dzieci jest porażenie mózgowe. Uszkodzenie mózgu może spowodować spastyczność kończyny dolnej, prowadząc do końskostopia.
W przypadku wrodzonej stopy końskiej wada ta ma charakter albo idiopatyczny (brak znanej przyczyny i wada niedziedziczna), albo nerwowo-mięśniowy, albo też jest związana z innymi wadami.
Częstość występowania
Ponad 100 000 noworodków na całym świecie rocznie cierpi z powodu końskostopia. We Francji częstość występowania wynosi od 1 do 2 przypadków na 1000 urodzeń, a choroba ta dotyka głównie chłopców (70% przypadków). W przypadku pacjentów w wieku powyżej 2 lat i starszych cierpiących na końskostopie z różnych powodów (porażenie mózgowe, udar mózgu, stwardnienie rozsiane, urazy mózgu itd.) dostępne dane nie pozwalają na oszacowanie częstości występowania tej choroby.
Aby zdiagnozować stopę końską w następstwie uszkodzenia mózgu, lekarz ocenia siłę mięśni kończyny przy rozciąganiu oraz wpływ spastyczności na wykonywanie codziennych czynności przez pacjenta.
Specjalista ocenia sytuację na trzech płaszczyznach:
- Diagnoza pozytywna: diagnoza polegająca na potwierdzeniu występowania stopy końskiej w przeciwieństwie do nieprawidłowego ułożenia stóp w macicy, które jest znacznie częstsze i znacznie łatwiej można je skorygować.
- Stopień nasilenia: diagnoza dotycząca oceny stopnia zaburzenia. Stopień nasilenia ogólnie określa się według klasyfikacji Diméglio-Bensahela. Ta czterostopniowa klasyfikacja jest kluczowa dla oceny prawdopodobnego przebiegu choroby i efektów terapii pacjenta.
- Diagnoza etiologiczna polegająca na określeniu przyczyn zaburzenia lub jego idiopatycznego charakteru.
Istnieje kilka metod leczenia mających na celu zmniejszenie lub złagodzenie końskiej stopy:
Leczenie końskiej stopy będącej wynikiem uszkodzenia mózgu:
- Iniekcje toksyny botulinowej typu A — toksyna botulinowa działa na poziomie połączenia nerwu z mięśniem, blokując tymczasowo transmisję neuroprzekaźnika (acetylocholiny). Wpływa na zmniejszenie napięcia mięśni spastycznych. Zastrzyki z toksyny botulinowej są stosowane w celu zmniejszenia spastyczności mięśnia trójgłowego łydki u pacjentów w wieku 2 lat i powyżej. Serie zastrzyków prowadzą do poprawy klinicznej w ciągu 15 dni i powinny być powtarzane (w zależności od potrzeb pacjenta) co 3 do 6 miesięcy.
- Leczenie farmakologiczne — leki zwiotczające mięśnie, takie jak baklofen, dantrolen i tyzanidyna, wykazują podobne działanie terapeutyczne jak toksyna botulinowa, jednak o innym profilu tolerancji. Rozluźniają spastyczne mięśnie nóg, pozwalając pacjentowi na postawienie stopy płasko na podłożu. Ten rodzaj leczenia powinien być regularnie oceniany w celu określenia jego skuteczności, dostosowania dawki oraz kontroli działań niepożądanych.
- Zastosowanie ortezy — pomaga utrzymać stopę w pozycji tworzącej kąt prosty z nogą, co ułatwia chodzenie.
Leczenie wrodzonej końskiej stopy:
- Leczenie czynnościowe — technika ta obejmuje codzienną rehabilitację prowadzoną przez wyspecjalizowanych fizjoterapeutów, jak również zastosowanie szyny i bandaży w celu utrzymania stopy w skorygowanej pozycji. Terapia musi być prowadzona przez prawie 3 lata.
-
Leczenie ortopedyczne metodą profesora Ponsetiego: na zdeformowaną stopę nakłada się w kilku etapach opatrunki gipsowe w celu skorygowania sztywności wiązadeł, rozciągnięcia połączeń mięśniowo-ścięgnistych oraz rozluźnienia hipertonicznych mięśni. Po korekcji wykonuje się w razie konieczności chirurgiczne wydłużenie ścięgna Achillesa (tenotomia) oraz stabilizuje się stopę po zabiegu za pomocą gipsów ortopedycznych.
- Leczenie chirurgiczne jest niekiedy konieczne w przypadku niepowodzenia leczenia końskiej stopy przy użyciu gipsu lub szyny. Zabieg chirurgiczny jest możliwy u pacjentów w wieku od 8 miesięcy i polega na wydłużeniu skurczonych struktur, które uniemożliwiają korektę deformacji (więzadła, ścięgna itp.). Operacja wykonywana jest jednym lub dwoma nacięciami, a następnie stopa jest utrzymywana w pozycji za pomocą trzpienia oraz gipsu przez 45 dni.
- Stosowanie szyny do utrzymywania odpowiedniej postawy oraz ortezy do chodzenia jest przydatne zarówno po zabiegach chirurgicznych, jak i u pacjentów nieoperowanych. Szyny i ortezy pomagają ustabilizować pozycję stopy i ułatwiają chodzenie.
Mimo skuteczności tych zabiegów, niektóre pozostałości wady są niemożliwe do uniknięcia u pacjentów z wrodzonym końskostopiem:
- mniejszy rozmiar stopy (1, 2, a nawet 3 rozmiary różnicy);
- szczuplejsza łydka dotkniętej stopy;
- osłabienie mięśni łydki.
Zniekształcenie typu stopa końska może być mniej lub bardziej poważne
Główną przyczyną stopy końskiej u osób dorosłych jest udar, a u dzieci — porażenie mózgowe
Ponad 100 000 noworodków na całym świecie rocznie cierpi z powodu końskostopia
Spastyczność
Spastyczność może być wywołana wieloma chorobami, takimi jak zaburzenia mózgowo-naczyniowe (udar mózgu), uraz mózgu, stwardnienie rozsiane, mózgowe porażenie dziecięce itp. Stale przykurczone kończyny tracą elastyczność, co może powodować ból oraz problemy z poruszaniem się. Firma Ipsen od lat zajmuje się rozwojem terapii spastyczności. Dowiedz się więcej na temat spastyczności, jej przyczyn i objawów, a także sposobów rozpoznawania oraz różnych metod leczenia.
Definicja
Spastyczność jest wzmożonym napięciem mięśniowym, narastającym pod wpływem szybkości ruchu. Tego typu napięcie mięśni wynika z braku hamowania odruchu rozciągowego (odruchu, który kurczy mięsień w odpowiedzi na jego rozciąganie), na przykład po urazie mózgu lub rdzenia kręgowego. Termin „spastyczność” odnosi się do zespołu czynności mięśniowych będących skutkiem uszkodzenia ośrodkowego układu nerwowego.
Zwiększone napięcie mięśniowe może być bardzo obciążające pacjenta i prowadzić do ciągłego skurczu, zbyt silnego w stanie spoczynku oraz zbyt intensywnego przy najmniejszej stymulacji. Zgięta kończyna nie może zostać wyprostowana, co powoduje problemy ruchowe oraz ból.
Objawy i skutki zdrowotne
Spastyczność rozwija się stopniowo i zazwyczaj do jej pojawienia się mija kilka tygodni (często od 1 do 3 miesięcy). Zawsze dotyczy grupy mniej lub bardziej znaczących mięśni, których lokalizacja odpowiada uszkodzeniu danego obszaru mózgu lub rdzenia kręgowego:
- lokalizacja ogniska udaru lub uszkodzenia mózgu w wyniku urazu: z reguły dotknięta jest jedna strona mózgu, co powoduje paraliż (częściowy, zwany niedowładem, lub pełny) po przeciwnej stronie ciała. Spastyczność może zatem dotyczyć całej połowy ciała, kończyny górnej lub dolnej, albo obu kończyn równocześnie..
- lokalizacja w przypadku stwardnienia rozsianego (SM): w przypadku SM zmiany mogą dotyczyć mózgu i/lub rdzenia kręgowego. Spastyczność może dotyczyć obydwu kończyn dolnych i skutkować trudnościami w poruszaniu się.
Spastyczność przejawia się podczas badania oporem w trakcie wykonywania ruchu biernego w stawie z jego przełamaniem w drugiej fazie ruchu oraz wzmożonym napięciem mięśniowym, dającym charakterystyczny opór podczas rozciągania mięśni.
Powikłania są zróżnicowane: w przypadku dużej spastyczności z czasem pojawiają się zmiany w strukturze mięśni (skurcze mięśni lub sztywność) i trudno je odróżnić od samej spastyczności. Mogą również pojawić się problemy związane z motoryką i równowagą, drżenie, ból, wpływ na wzrost u dzieci lub na jakość życia pacjentów.
Etiologia spastyczności
Spastyczność może przejawiać się w różnych formach, towarzysząc wielu schorzeniom neurologicznym i rozwijając się w wyniku:
- udaru mózgu,
- urazów mózgu,
- uszkodzeń rdzenia kręgowego,
- stwardnienia rozsianego,
- porażenia mózgowego.
Najczęstszą przyczyną spastyczności jest udar mózgu. Z powodu zablokowania tętnicy (udar niedokrwienny) lub jej pęknięcia (udar krwotoczny), krew nie może zostać dostarczona do komórek w części mózgu, co powoduje jego uszkodzenie. Spastyczność rozwija się stopniowo, najczęściej między 1 a 3 miesiącami po udarze mózgu i dotyka do około 40% pacjentów.
W celu postawienia rozpoznania konieczna jest dokładna analiza kliniczna by określić nasilenie, skutki oraz rodzaj spastyczności. Aby postawić rozpoznanie, lekarz ocenia:
- nasilenie spastyczności – do tego służą odpowiednie testy
- wpływ spastyczności na codzienne życie pacjenta. Spastyczność jest leczona jedynie wtedy, gdy wywołuje upośledzenie czynnościowe, które może być zmniejszone poprzez to leczenie.
- Spastyczność jest potencjalnie „przydatna”, zwłaszcza gdy pomaga równoważyć inny deficyt neurologiczny (napięcie mięśni umożliwia, na przykład utrzymanie pozycji stojącej mimo osłabienia lub niedowładu — częściowego porażenia — nogi). Zmniejszenie spastyczności w tym przypadku ma negatywny wpływ na układ ruchowy pacjenta.
W przypadku leczenia, jego strategia opiera się na zindywidualizowanym podejściu do pacjenta i ma na celu:
- poprawę zdolności motorycznych (wykonanie danego ruchu lub chodzenie);
- zmniejszenie bólu;
- poprawę opieki pielęgniarskiej nad pacjentem.
Istnieje kilka rodzajów leczenia:
- Iniekcje toksyny botulinowej typu A, która działa na poziomie połączeń nerwowo-mięśniowych w mięśniu docelowym poprzez hamowanie wydzielania acetylocholiny (neuroprzekaźnika), co pomaga ograniczyć skurcze mięśni.
- Leczenie farmakologiczne: na ogół stosowane w przypadku wyraźnej spastyczności, opiera się na podawaniu środków zwiotczających mięśnie, takich jak baklofen, dantrolen i tyzanidyna. Pochodne konopi, podawane doustnie, wykazują pewną skuteczność w zakresie spastyczności, zwłaszcza gdy jej przyczyną jest stwardnienie rozsiane. Ten rodzaj leczenia powinien być regularnie oceniany w celu określenia jego skuteczności, dostosowania dawki oraz kontroli działań niepożądanych. W leczeniu ciężkiej spastyczności w przypadku porażenia mózgowego, pourazowych uszkodzeń rdzenia kręgowego lub uszkodzeń wtórnych w wyniku stwardnienia rozsianego, po niepowodzeniu podawania doustnego lub gdy skuteczne dawki powodują działania niepożądane w obrębie ośrodkowego układu nerwowego, można rozważyć dokanałowe podawanie baklofenu.
- Leczenie chirurgiczne jest podejmowane w przypadku nieskuteczności innych metod leczenia.
- Fizjoterapia: niezbędna nawet w przypadku leczenia farmakologicznego lub chirurgicznego, pomaga pacjentowi nauczyć się, jak najlepiej wykorzystać pozostałe zdolności motoryczne. Prawidłowo dobrane ćwiczenia rozciągające pozwalają utrzymać większy zakres ruchu w stawach i zapobiegają powstawaniu przykurczy mięśniowych oraz czasami bolesnej sztywności mięśni.
- Zastosowanie szyn oraz ortez zalecanych przez ortopedę, neurologa lub fizjoterapeutę. Urządzenia te stosuje się w celu utrzymania spastycznej kończyny w ustalonym położeniu.
Spastyczność rozwija się stopniowo
W celu postawienia diagnozy konieczne jest dokładne badanie neurologiczne
Nie każdy chory na spastyczność wymaga leczenia
Źródło:
„Toksyna botulinowa w praktyce neurologicznej” pod redakcją Jarosława Sławka i Moniki Rudzińskiej, tom 1. Via Medica, Gdańsk 2015
Lokalne badania kliniczne
Odkryj nasze badania kliniczne
Jakość życia pacjentów z akromegaliã leczonych lanreotydem
Ocena jakości życia w populacji pacjentów leczonych lanreotydem 120 mg w ciągu 24 miesięcznej obserwacji
Badanie obserwacyjne na terenie Rzeczpospolitej Polskiej
Zgoda Komisji Etycznej z dnia 24.10.2014r przyjęta Uchwała nr 127/14
Rejestracja na stronie clinicaltrials.gov ID: A-38-52030-306
Liczba ośrodków badawczych: 30
Liczba pacjentów: 150
Consumer healthcare
Consumer healthcare zajmuje się podstawową opieką medyczną.
Biegunka
Biegunka oznacza oddawanie stolców płynnych, miękkich i/lub zbyt częstych, co powoduje poważne utrudnienia w wykonywaniu codziennych czynności u dorosłych oraz, szczególnie u małych dzieci, ryzyko odwodnienia, które może być potencjalnie niebezpieczne w razie niepodjęcia odpowiedniego leczenia. Dowiedz się więcej na temat tego zaburzenia, jego diagnozowania i różnych metod leczenia.
Definicja
Biegunka występuje w przypadku codziennego oddawania częstych stolców o zbyt miękkiej lub płynnej konsystencji (3 lub więcej stolców dziennie).
Istnieją 4 główne mechanizmy powstawania biegunki:
- biegunka osmotyczna spowodowana obecnością w jelicie cienkim lub okrężnicy substancji, które zatrzymują wodę;
- biegunka wydzielnicza, której przyczyną jest zbyt duża utrata wody i elektrolitów w wyniku nadmiernej stymulacji wydzieliny jelitowej i/lub hamowania mechanizmów wchłaniania z jelita cienkiego lub okrężnicy;
- biegunki związane z zaburzeniami ruchowymi z przyspieszeniem pasażu jelitowego ograniczającym wchłanianie wody w jelicie grubym;
- biegunki związane ze zniszczeniem śluzówki odpowiedzialnym za zmiany w przepuszczalności jelit, zjawiska złego wchłaniania, wyciek elektrolitów i białek do światła jelita; w takiej sytuacji mówimy o czerwonce, charakteryzującej się obecnością krwi oraz śluzu w stolcu; ten typ mechanizmu jest najczęściej obserwowany w trakcie zakażenia mikroorganizmami, takimi jak bakterie i wirusy, które działają bezpośrednio lub za pośrednictwem toksyny.
Często u podstaw biegunki występuje kilka mechanizmów jednocześnie.
Objawy i skutki zdrowotne
Ostra biegunka (zbyt częste i wodniste stolce) jest najczęściej powiązana z innymi objawami:
- skurczowe bóle brzucha,
- gorączka,
- nudności lub wymioty.
Głównym ryzykiem związanym z wystąpieniem ostrej biegunki jest odwodnienie organizmu. Taka sytuacja może być poważna u dzieci i osób słabszych, w szczególności osób starszych i/lub cierpiących z powodu innej choroby przewlekłej.
Glównymi przycynami ostrej biegunka są:
- zakażenia wirusowe (rotawirus, norowirus, adenowirus), które mogą wystąpić w czasie zakażenia zbiorowego (w przypadku dzieci np. przedszkola, szkoły) lub rodzinnego;
- zatrucie pokarmowe związane z zanieczyszczeniem żywności lub napojów przez organizmy zakaźne lub toksyczne (Salmonella, Shigella, Escherichia coli, Campylobacter itp.),
- zmiana nawyków żywieniowych lub trybu odżywiania, zwłaszcza podczas podróży za granicą, „pożywne posiłki”;
- nietolerancja lub alergia pokarmowa (laktoza itp.);
- skutki uboczne niektórych leków (na przykład antybiotyków), które wpływają na florę jelitową, leczenia stosowanego w dnie moczanowej, cukrzycy itd.;
- nieswoiste choroby zapalne jelit (choroba Crohna, zapalenie jelita grubego) lub wewnątrzwydzielnicze (nadczynność tarczycy, cukrzyca);
- czynnościowe choroby jelit (zespół jelita drażliwego);
- niepokój, sytuacje stresowe (np. egzaminy).
Częstość występowania
Każdego roku występuje szczyt epidemii zimowej, zazwyczaj w grudniu i styczniu, w dużej mierze przypisywany głównie infekcjom wirusowym, zaś bardziej umiarkowany wzrost zachorowań obserwuje się latem, co jest związane z zakażeniami bakteryjnymi. Ryzyko poważnych powikłań lub zgonów wywołanych ostrą biegunką nawet w krajach wysoko rozwiniętych nie jest zerowe, choć jest niewspółmierne do ryzyka w krajach rozwijających się, i powinno zawsze być szczególnie traktowane u małych dzieci w wieku poniżej 5 lat oraz osób osłabionych.
Rozróżnia się 2 główne typy biegunki: biegunkę ostrą, o nagłym początku, trwającą z reguły od 3 do 7 dni, oraz biegunkę przewlekłą, utrzymującą się ponad 4 kolejne tygodnie lub nawet kilka miesięcy, głównie u osób dorosłych.
Można wyszczególnić także 3 główne rodzaje przyczyn biegunki: biegunka spowodowana chorobą jelit (na przykład chorobą zapalną), biegunka pochodzenia czynnościowego, która może występować na przykład w przebiegu jelita drażliwego, oraz biegunka spowodowana zakażeniem wirusowym lub bakteryjnym, zwykle o ostrym początku.
W przypadku ostrej biegunki należy przede wszystkim rozważyć zapobieganie odwodnieniu i odpowiednie dostosowanie diety:
- Nawadnianie organizmu wodą pitną, wodą mineralną butelkowaną lub ewentualnie wodą przegotowaną.
- W przypadku ostrej biegunki u dzieci i osób osłabionych zaleca się stosowanie doustnych roztworów nawadniających zawierających elektrolity, środki alkalizujące oraz cukry (węglowodany), przeznaczonych do natychmiastowego zapobiegania odwodnieniu organizmu.
- Małe częste posiłki składające się z potraw gotowanych i ubogich w błonnik (ryż, makaron, gotowana marchew), słone zupy, białko jaj, białe mięso (kurczak), ryby. Należy unikać alkoholu, kawy i herbaty, które mogą przyspieszać pasaż jelitowy, napojów gazowanych oraz innych napojów o wysokiej zawartości słodzików (sorbitol), potraw tłustych, smażonych, fasoli, soczewicy oraz produktów wysokobłonnikowych.
Objawowe leczenie przeciwbiegunkowe, które wiąże toksyny i bakterie oraz chroni uszkodzoną błonę śluzową przewodu pokarmowego, może sprzyjać przywróceniu prawidłowego pasażu jelitowego oraz prawidłowego wchłaniania i wydzielania w jelitach.
3
wypróżnienia dziennie lub więcej, stolce o płynnej lub miękkiej konsystencj
Często powiązana z
Bólem brzucha, nudnościami lub wymiotami oraz gorączką
Dzieci poniżej 4. roku życia
są najbardziej narażone na ryzyko wystąpienia powikłań, zwłaszcza odwodnienie może być u nich nawet śmiertelne
Zaparcia
Zaparcie to zbyt rzadkie lub utrudnione oddawanie stolca. Dowiedz się więcej na temat tego zaburzenia, jego diagnozowania i różnych metod leczenia.
Definicja
O zaparciach mówimy wtedy, gdy liczba wypróżnień jest mniejsza niż trzy razy na tydzień lub konsystencja stolca jest zbyt twarda albo stolec jest małych rozmiarów i występują trudności w jego oddawaniu. W takiej sytuacji oddawanie stolca może wymagać wysiłku, czasem bolesnego, związanego na ogół z uczuciem niepełnego wypróżnienia, bólami brzucha i wzdęciami.
Zaparcia mogą mieć dwa główne mechanizmy: spowolniony pasaż jelitowy oraz zaburzenia mechanizmu wypróżniania stolca na poziomie odbytnicy i odbytu.
Objawy i skutki zdrowotne
Zaparcia charakteryzują się wystąpieniem wielu objawów:
- zmniejszenie częstotliwości wypróżnień do mniej niż trzy razy w tygodniu;
- stolce twarde i skąpe, związane z uczuciem niepełnego wypróżnienia;
- trudności w wypróżnieniu (defekacji), a nawet ból;
- uczucie dyskomfortu w jamie brzusznej, skurcze i wzdęcia.
Przyczyny i czynniki ryzyka
Wyróżnia się kilka przyczyn występowania zaparć:
- zmiana codziennych nawyków żywieniowych — modyfikacja diety (zwłaszcza podczas podróży za granicą);
- brak równowagi w diecie, dieta uboga w błonnik pokarmowy;
- zaburzenia wypróżnień, które mogą być spowodowane zbyt wczesną lub zbyt surową nauką załatwiania potrzeb fizjologicznych w dzieciństwie, brakiem dostępu do toalety (brakiem prywatności w miejscu publicznym, stresującą pracą zawodową itp.), prowadzą do nieprawidłowego odruchu wydalania stolca;
- skutki uboczne niektórych leków (w tym przeciwkaszlowych, niektórych leków przeciwbólowych, takich jak kodeina, niektórych leków przeciwdepresyjnych itd.);
- siedzący tryb życia, zwłaszcza u osób starszych i niepełnosprawnych;
- choroby jelit również mogą być przyczyną zaparć, jednak znacznie rzadziej (rak jelita grubego itp.), a także choroby przewlekłe (cukrzyca, niedoczynność tarczycy, choroba Parkinsona, stwardnienie rozsiane itd.);
- zazwyczaj zaparcia maja charakter czynnościowy, a przyczyna ich występowania jest nieznana.
Częstość występowania
Zaparcia czynnościowe dotyczą od 1% do 20% osób dorosłych w krajach zachodnich, i występują częściej wśród kobiet (2/3) niż wśród mężczyzn (1/3). W badaniach przeprowadzonych u osób starszych, aż 50% osób przebywających w placówkach opiekuńczych miało zaparcia, w szczególności ze względu na siedzący tryb życia, stosowanie dużej ilości leków i choroby związane z podeszłym wiekiem.
Zaparcia u dzieci pozostają często nierozpoznane, zazwyczaj objawiają się nawracającymi bólami brzucha.
Normalna częstotliwość oddawania stolca może wahać się u różnych osób od trzech razy dziennie do trzech razy w tygodniu. Zaparcia definiuje się jako nieprawidłowe zmniejszenie częstotliwości oddawania stolca (mniej niż trzy razy w tygodniu) lub oddawanie stolca o zbyt twardej konsystencji, a także jako trudności w wypróżnianiu. Zaparcia przewlekłe występują wtedy, gdy objawy utrzymują się dłużej niż sześć miesięcy.
W przypadku zaparć należy najpierw skonsultować się z lekarzem, po to aby wykluczyć chorobę mogącą być przyczyną ich wystąpienia.
W przypadku zaparć pochodzenia czynnościowego, najważniejszą zasadą jest dostosowanie swojej diety oraz trybu życia:
- codzienna spokojna wizyta w toalecie, jeśli to możliwe o stałej porze i jak tylko zajdzie taka potrzeba;
- stosowanie zrównoważonej, bogatej w błonnik diety: zielone warzywa (brukselka, brokuły), rośliny strączkowe (biała fasola, groch, soczewica), owoce (jabłka, gruszki, owoce suszone, śliwki, figi, daktyle), produkty z pełnego ziarna (pszenica, otręby pszenne, bulgur, kukurydza, orkisz, siemię lniane);
- utrzymywanie odpowiedniego nawodnienia; pomocne może być także picie bogatej w magnez wody mineralnej.
Jednak czasami konieczne staje się zastosowanie dodatkowo leku przeczyszczającego:
- osmotyczne środki przeczyszczające zmiękczają i zwiększają objętość stolca poprzez zwiększenie zawartości wody;
- balastowe środki przeczyszczające zmieniają objętość i konsystencję stolca;
- nawilżające środki przeczyszczające ułatwiają pasaż stolca w jelitach;
- stymulujące środki przeczyszczające przyspieszają ruchy jelit;
- środki przeczyszczające stosowane doodbytniczo (czopki, lewatywy) wpływają na wyzwalanie refleksu do defekacji.
Mniej niż 3 wypróżnienia
W tygodniu lub zbyt twarde stolce oraz trudności w wypróżnieniu
Od 10 do 30%
Problem dotyczy od 10 do 30% dorosłych, występuje częściej u kobiet
Zaparcia u dzieci
Często pozostają nierozpoznane